Human fetal membranes generate distinct cytokine profiles in response to bacterial Toll-like receptor and nod-like receptor agonists.
Hoang, M; Potter, JA; Gysler, SM; Han, CS; Guller, S; Norwitz, ER; Abrahams, VM
Biology of reproduction
90
39
2014
Abstract anzeigen
Bacterial infection-associated inflammation is thought to be a major cause of preterm premature rupture of membranes. Proinflammatory cytokines, such as interleukin 1B (IL1B), can weaken fetal membranes (FM) by upregulating matrix metalloproteinases and inducing apoptosis. The mechanism by which infection leads to inflammation at the maternal-fetal interface and subsequent preterm birth is thought to involve innate immune pattern recognition receptors (PRR), such as the Toll-like receptors (TLR) and Nod-like receptors (NLR), which recognize pathogen-associated molecular patterns (PAMPs). The objective of this study was to determine the cytokine profile generated by FMs in response to the bacterial TLR and NLR agonists peptidoglycan (PDG; TLR2), lipopolysaccharide (LPS; TLR4), flagellin (TLR5), CpG ODN (TLR9), iE-DAP (Nod1), and MDP (Nod2). PDG, LPS, flagellin, iE-DAP, and MDP triggered FMs to generate an inflammatory response, but the cytokine profiles were distinct for each TLR and NLR agonist, and only IL1B and RANTES were commonly upregulated in response to all five PAMPs. CpG ODN, in contrast, had a mild stimulatory effect only on MCP-1 and primarily downregulated basal FM cytokine production. IL1B secretion induced by PDG, LPS, flagellin, iE-DAP, and MDP was associated with its processing. Furthermore, FM IL1B secretion in response to TLR2, TLR4, and TLR5 activation was caspase 1-dependent, whereas Nod1 and Nod2 induced IL1B secretion independent of caspase 1. These findings demonstrate that FMs respond to different bacterial TLR and NLR PAMPs by generating distinct inflammatory cytokine profiles through distinct mechanisms that are specific to the innate immune PRR activated. | | 24429216
 |
Cyclic-di-GMP and cyclic-di-AMP activate the NLRP3 inflammasome.
Abdul-Sater, AA; Tattoli, I; Jin, L; Grajkowski, A; Levi, A; Koller, BH; Allen, IC; Beaucage, SL; Fitzgerald, KA; Ting, JP; Cambier, JC; Girardin, SE; Schindler, C
EMBO reports
14
900-6
2013
Abstract anzeigen
The cyclic dinucleotides 3'-5'diadenylate (c-diAMP) and 3'-5' diguanylate (c-diGMP) are important bacterial second messengers that have recently been shown to stimulate the secretion of type I Interferons (IFN-Is) through the c-diGMP-binding protein MPYS/STING. Here, we show that physiologically relevant levels of cyclic dinucleotides also stimulate a robust secretion of IL-1β through the NLRP3 inflammasome. Intriguingly, this response is independent of MPYS/STING. Consistent with most NLRP3 inflammasome activators, the response to c-diGMP is dependent on the mobilization of potassium and calcium ions. However, in contrast to other NLRP3 inflammasome activators, this response is not associated with significant changes in mitochondrial potential or the generation of mitochondrial reactive oxygen species. Thus, cyclic dinucleotides activate the NLRP3 inflammasome through a unique pathway that could have evolved to detect pervasive bacterial pathogen-associated molecular patterns associated with intracellular infections. | | 24008845
|
Caspase-1 promotes Epstein-Barr virus replication by targeting the large tegument protein deneddylase to the nucleus of productively infected cells.
Gastaldello, S; Chen, X; Callegari, S; Masucci, MG
PLoS pathogens
9
e1003664
2013
Abstract anzeigen
The large tegument proteins of herpesviruses contain N-terminal cysteine proteases with potent ubiquitin and NEDD8-specific deconjugase activities, but the function of the enzymes during virus replication remains largely unknown. Using as model BPLF1, the homologue encoded by Epstein-Barr virus (EBV), we found that induction of the productive virus cycle does not affect the total level of ubiquitin-conjugation but is accompanied by a BPLF1-dependent decrease of NEDD8-adducts and accumulation of free NEDD8. Expression of BPLF1 promotes cullin degradation and the stabilization of cullin-RING ligases (CRLs) substrates in the nucleus, while cytoplasmic CRLs and their substrates are not affected. The inactivation of nuclear CRLs is reversed by the N-terminus of CAND1, which inhibits the binding of BPLF1 to cullins and prevents efficient viral DNA replication. Targeting of the deneddylase activity to the nucleus is dependent on processing of the catalytic N-terminus by caspase-1. Inhibition of caspase-1 severely impairs viral DNA synthesis and the release of infectious virus, pointing a previously unrecognized role of the cellular response to danger signals triggered by EBV reactivation in promoting virus replication. | | 24130483
|
Nod1, but not the ASC inflammasome, contributes to induction of IL-1β secretion in human trophoblasts after sensing of Chlamydia trachomatis.
Kavathas, PB; Boeras, CM; Mulla, MJ; Abrahams, VM
Mucosal immunology
6
235-43
2013
Abstract anzeigen
Chlamydia trachomatis (Ct) is an obligate intracellular bacterial pathogen. Previously, we showed that infection of human trophoblast cells by Ct triggers the secretion of the pro-inflammatory cytokine, interleukin (IL)-1β. The aim of this study was to understand the innate immune pathways involved in trophoblast production of IL-1β after Ct infection. The approach we took was to inhibit the expression or function of the key Toll-like receptors (TLRs), Nod-like receptors, and inflammasome components that have been associated with chlamydia infection. In this study, we report that Ct-induced trophoblast IL-1β secretion is associated with the transcription of IL-1β mRNA, the translation and processing of pro-IL-1β, and the activation of caspase-1. In addition, we demonstrate that Ct-induced IL-1β production and secretion by the trophoblast is independent of TLR2, TLR4, MyD88, and the Nalp3/ASC inflammasome. Instead we report, for the first time, the importance of Nod1 for mediating trophoblast IL-1β secretion in response to a Ct infection. | | 22763410
|
The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP.
Lee, GS; Subramanian, N; Kim, AI; Aksentijevich, I; Goldbach-Mansky, R; Sacks, DB; Germain, RN; Kastner, DL; Chae, JJ
Nature
492
123-7
2011
Abstract anzeigen
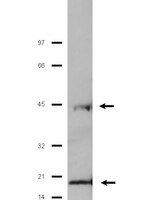
Mutations in the gene encoding NLRP3 cause a spectrum of autoinflammatory diseases known as cryopyrin-associated periodic syndromes (CAPS). NLRP3 is a key component of one of several distinct cytoplasmic multiprotein complexes (inflammasomes) that mediate the maturation of the proinflammatory cytokine interleukin-1β (IL-1β) by activating caspase-1. Although several models for inflammasome activation, such as K(+) efflux, generation of reactive oxygen species and lysosomal destabilization, have been proposed, the precise molecular mechanism of NLRP3 inflammasome activation, as well as the mechanism by which CAPS-associated mutations activate NLRP3, remain to be elucidated. Here we show that the murine calcium-sensing receptor (CASR) activates the NLRP3 inflammasome, mediated by increased intracellular Ca(2+) and decreased cellular cyclic AMP (cAMP). Ca(2+) or other CASR agonists activate the NLRP3 inflammasome in the absence of exogenous ATP, whereas knockdown of CASR reduces inflammasome activation in response to known NLRP3 activators. CASR activates the NLRP3 inflammasome through phospholipase C, which catalyses inositol-1,4,5-trisphosphate production and thereby induces release of Ca(2+) from endoplasmic reticulum stores. The increased cytoplasmic Ca(2+) promotes the assembly of inflammasome components, and intracellular Ca(2+) is required for spontaneous inflammasome activity in cells from patients with CAPS. CASR stimulation also results in reduced intracellular cAMP, which independently activates the NLRP3 inflammasome. cAMP binds to NLRP3 directly to inhibit inflammasome assembly, and downregulation of cAMP relieves this inhibition. The binding affinity of cAMP for CAPS-associated mutant NLRP3 is substantially lower than for wild-type NLRP3, and the uncontrolled mature IL-1β production from CAPS patients' peripheral blood mononuclear cells is attenuated by increasing cAMP. Taken together, these findings indicate that Ca(2+) and cAMP are two key molecular regulators of the NLRP3 inflammasome that have critical roles in the molecular pathogenesis of CAPS. | Western Blotting | 23143333
|
TANK-binding kinase 1 (TBK1) controls cell survival through PAI-2/serpinB2 and transglutaminase 2.
Delhase, M; Kim, SY; Lee, H; Naiki-Ito, A; Chen, Y; Ahn, ER; Murata, K; Kim, SJ; Lautsch, N; Kobayashi, KS; Shirai, T; Karin, M; Nakanishi, M
Proceedings of the National Academy of Sciences of the United States of America
109
E177-86
2011
Abstract anzeigen
The decision between survival and death in cells exposed to TNF relies on a highly regulated equilibrium between proapoptotic and antiapoptotic factors. The TNF-activated antiapoptotic response depends on several transcription factors, including NF-κB and its RelA/p65 subunit, that are activated through phosphorylation-mediated degradation of IκB inhibitors, a process controlled by the IκB kinase complex. Genetic studies in mice have identified the IκB kinase-related kinase TANK-binding kinase 1 (TBK1; also called NAK or T2K) as an additional regulatory molecule that promotes survival downstream of TNF, but the mechanism through which TBK1 exerts its survival function has remained elusive. Here we show that TBK1 triggers an antiapoptotic response by controlling a specific RelA/p65 phosphorylation event. TBK1-induced RelA phosphorylation results in inducible expression of plasminogen activator inhibitor-2 (PAI-2), a member of the serpin family with known antiapoptotic activity. PAI-2 limits caspase-3 activation through stabilization of transglutaminase 2 (TG2), which cross-links and inactivates procaspase-3. Importantly, Tg2(-/-) mice were found to be more susceptible to apoptotic cell death in two models of TNF-dependent acute liver injury. Our results establish PAI-2 and TG2 as downstream mediators in the antiapoptotic response triggered upon TBK1 activation. | | 22203995
|
Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis.
Dixon, LJ; Berk, M; Thapaliya, S; Papouchado, BG; Feldstein, AE
Laboratory investigation; a journal of technical methods and pathology
92
713-23
2011
Abstract anzeigen
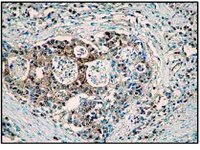
Non-alcoholic steatohepatitis (NASH) is typically associated with pro-apoptotic caspase activation. A potential role for pro-inflammatory caspases remains incompletely understood. Our aims were to examine a potential role of caspase-1 in the development of liver damage and fibrosis in NASH. C57BL/6 wild type (WT) developed marked steatohepatitis, activation, fibrosis and increased hepatic caspase-1 and interleukin-1β expression when placed on the methionine- and choline-deficient (MCD) diet. Marked caspase-1 activation was detected in the liver of MCD-fed mice. Hepatocyte and non-parenchymal fractionation of the livers further demonstrated that caspase-1 activation after MCD feeding was mainly localized to non-parenchymal cells. Caspase-1-knockout (Casp1(-/-)) mice on the MCD diet showed marked reduction in mRNA expression of genes involved in inflammation and fibrogenesis (tumor necrosis factor-α was 7.6-fold greater in WT vs Casp1(-/-) MCD-fed mice; F4/80 was 1.5-fold greater in WT vs Casp1(-/-) MCD-fed mice; α-smooth muscle actin was 3.2-fold greater in WT vs Casp1(-/-) MCD-fed mice; collagen 1-α was 7.6-fold greater in WT vs Casp1(-/-) MCD-fed mice; transforming growth factor-β was 2.4-fold greater in WT vs Casp1(-/-) MCD-fed mice; cysteine- and glycine-rich protein 2 was 3.2-fold greater in WT vs Casp1(-/-) MCD-fed mice). Furthermore, Sirius red staining for hepatic collagen deposition was significantly reduced in Casp1(-/-) MCD-fed mice compared with WT MCD-fed animals. However, serum alanine aminotransferase levels, caspase-3 activity and terminal deoxynucleotidyl transferase dUTP nick-end labeling-positive cells were similar in Casp1(-/-) and WT mice on the MCD diet. Selective Kupffer cell depletion by clodronate injection markedly suppressed MCD-induced caspase-1 activation and protected mice from fibrogenesis and fibrosis associated with this diet. The conclusion of this study is that it uncovers a novel role for caspase-1 in inflammation and fibrosis during NASH development. | Immunohistochemistry | 22411067
|
Uric acid induces trophoblast IL-1β production via the inflammasome: implications for the pathogenesis of preeclampsia.
Mulla, MJ; Myrtolli, K; Potter, J; Boeras, C; Kavathas, PB; Sfakianaki, AK; Tadesse, S; Norwitz, ER; Guller, S; Abrahams, VM
American journal of reproductive immunology (New York, N.Y. : 1989)
65
542-8
2010
Abstract anzeigen
Preeclampsia is associated with hyperuricemia, which correlates with the disease severity. Levels of circulating uric acid increase before the clinical manifestations, suggesting that they may be causally related. Uric acid, or monosodium urate (MSU), activates the Nod-like receptor, Nalp3, leading to inflammasome activation and IL-1β processing. Because preeclampsia is associated with placental immune⁄ inflammatory dysregulation, we sought to determine in the trophoblast, the presence of the Nalp3 inflammasome, and the effect of MSU on its activation.Isolated first- and third-trimester trophoblasts were assessed for expression of the inflammasome components, Nalp1, Nalp3, and ASC. First-trimester trophoblast cells were incubated with or without MSU, and after which, IL-1β secretion and processing and caspase-1 activation were determined.Trophoblast cells expressed Nalp1, Nalp3, and ASC under basal conditions. Following incubation with MSU, first-trimester trophoblast IL-1β secretion was upregulated. This correlated with increased expression levels of active IL-1β and active caspase-1. ASC knockdown reduced MSU-induced IL-1β secretion.These findings demonstrate that uric acid activates the inflammasome in the trophoblast, leading to IL-1β production. This may provide a novel mechanism for the induction of inflammation at the maternal–fetal interface leading to placental dysfunction and adverse pregnancy outcome, including preeclampsia. | | 21352397
|
CONSTITUTIONAL METHYLATION OF THE BRCA1 PROMOTER IS SPECIFICALLY ASSOCIATED WITH BRCA1 MUTATION-ASSOCIATED PATHOLOGY IN EARLY-ONSET BREAST CANCER.
Wong EM, Southey MC, Fox SB, Brown MA, Dowty JG, Jenkins MA, Giles GG, Hopper J, Dobrovic A
Cancer Prev Res (Phila)
2009
Abstract anzeigen
Women carrying germline mutations in BRCA1 are at a substantially elevated risk of breast cancer and their tumors typically have distinctive morphological features. We hypothesised that constitutional methylation of the BRCA1 promoter region could give rise to such breast cancers in women. We selected 255 women diagnosed with breast cancer before the age of 40 years for whom BRCA1 germline mutations had not been identified. 52 had five or more of nine BRCA1 mutation-associated morphological features (group 1), 39 had four (group 2), and 164 had three or less (group 3). The prevalence of detectable BRCA1 promoter methylation in peripheral blood DNA decreased from 31% to 10% to 5% across groups 1-3, respectively (p=0.000002) and was significantly greater than the 4% frequency in unaffected controls (p=0.004). Peripheral blood methylation was associated with a 3.5-fold (95% CI 1.4 - 10.5) increased risk of having early onset breast cancer. Methylation was consistently mosaic in the peripheral blood where the estimated allelic frequency of BRCA1 promoter methylation ranged from 0.1% to 17%. Group 1 women but not group 3 women with detectable methylation of peripheral blood DNA had high levels of BRCA1 promoter methylation of their tumor DNA, indicating that constitutional BRCA1 methylation strongly predisposes towards the development of BRCA1 methylated tumors that then have features resembling BRCA1 mutated tumors. Screening peripheral blood for BRCA1 promoter methylation might thus predict early-onset breast cancers. This raises the possibility of chemoprevention or other intervention to diminish the risk of developing breast cancer in these women. | | 20978112
|
Caspase-1 contributes to Chlamydia trachomatis-induced upper urogenital tract inflammatory pathologies without affecting the course of infection.
Cheng, W; Shivshankar, P; Li, Z; Chen, L; Yeh, IT; Zhong, G
Infection and immunity
76
515-22
2008
Abstract anzeigen
Chlamydia trachomatis infection induces inflammatory pathologies in the upper genital tract, potentially leading to ectopic pregnancy and infertility in the affected women. Caspase-1 is required for processing and release of the inflammatory cytokines interleukin-1beta (IL-1beta), IL-18, and possibly IL-33. In the present study, we evaluated the role of caspase-1 in chlamydial infection and pathogenesis. Although chlamydial infection induced caspase-1 activation and processing of IL-1beta, mice competent and mice deficient in caspase-1 experienced similar courses of chlamydial infection in their urogenital tracts, suggesting that Chlamydia-activated caspase-1 did not play a significant role in resolution of chlamydial infection. However, when genital tract tissue pathologies were examined, the caspase-1-deficient mice displayed much reduced inflammatory damage. The reduction in inflammation was most obvious in the fallopian tube tissue. These observations demonstrated that although caspase-1 is not required for controlling chlamydial infection, caspase-1-mediated responses can exacerbate the Chlamydia-induced inflammatory pathologies in the upper genital tract, suggesting that the host caspase-1 may be targeted for selectively attenuating chlamydial pathogenicity without affecting the host defense against chlamydial infection. | | 18025098
|