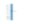A cellular model of amyloid precursor protein processing and amyloid-β peptide production.
Macias, MP; Gonzales, AM; Siniard, AL; Walker, AW; Corneveaux, JJ; Huentelman, MJ; Sabbagh, MN; Decourt, B
Journal of neuroscience methods
223
114-22
2014
Mostrar Resumo
A hallmark pathologic feature of Alzheimer's disease (AD) is accumulation of neuritic senile plaques in the brain parenchyma. Neurotoxic plaque cores are composed predominantly of amyloid-β (Aβ) peptides of 40 and 42 amino acids in length, formed by sequential cleavage of amyloid precursor protein (APP) by β-, and γ-secretases. There is a great interest in approaches to modulate Aβ peptide production and develop therapeutic interventions to reduce Aβ levels to halt or slow the progression of neurodegeneration.We characterized and present the BE(2)-M17 human neuroblastoma cell line as a novel in vitro model of the APP-cleavage cascade to support future (1) functional studies of molecular regulators in Aβ production, and (2) high-throughput screening assays of new pharmacotherapeutics.In BE(2)-M17 cells, both RNA (i.e., RT-PCR, RNA sequencing) and protein analyses (i.e., Western blots, ELISA), show endogenous expression of critical components of the amyloidogenic pathway, APP-cleavage intermediates CTF83 and CTF99, and final cleavage products Aβ40 and Aβ42. We further report effects of retinoic acid-mediated differentiation on morphology and gene expression in this cell line.In contrast to primary isolates or other cell lines reported in current literature, BE(2)-M17 not only sustains baseline expression of the full contingent of APP-processing components, but also remains stably adherent during culture, facilitating experimental manipulations.Our evidence supports the use of BE(2)-M17 as a novel, human, cell-based model of the APP processing pathway that offers a potential streamlined approach to dissect molecular functions of endogenous regulatory pathways, and perform mechanistic studies to identify modulators of Aβ production. | | 24333289
 |
Inhibition of amyloid precursor protein secretases reduces recovery after spinal cord injury.
Pajoohesh-Ganji, A; Burns, MP; Pal-Ghosh, S; Tadvalkar, G; Hokenbury, NG; Stepp, MA; Faden, AI
Brain research
1560
73-82
2014
Mostrar Resumo
Amyloid-β (Aβ) is produced through the enzymatic cleavage of amyloid precursor protein (APP) by β (Bace1) and γ-secretases. The accumulation and aggregation of Aβ as amyloid plaques is the hallmark pathology of Alzheimer׳s disease and has been found in other neurological disorders, such as traumatic brain injury and multiple sclerosis. Although the role of Aβ after injury is not well understood, several studies have reported a negative correlation between Aβ formation and functional outcome. In this study we show that levels of APP, the enzymes cleaving APP (Bace1 and γ-secretase), and Aβ are significantly increased from 1 to 3 days after impact spinal cord injury (SCI) in mice. To determine the role of Aβ after SCI, we reduced or inhibited Aβ in vivo through pharmacological (using DAPT) or genetic (Bace1 knockout mice) approaches. We found that these interventions significantly impaired functional recovery as evaluated by white matter sparing and behavioral testing. These data are consistent with a beneficial role for Aβ after SCI. | | 24630972
|
Distinct patterns of APP processing in the CNS in autosomal-dominant and sporadic Alzheimer disease.
Pera, M; Alcolea, D; Sánchez-Valle, R; Guardia-Laguarta, C; Colom-Cadena, M; Badiola, N; Suárez-Calvet, M; Lladó, A; Barrera-Ocampo, AA; Sepulveda-Falla, D; Blesa, R; Molinuevo, JL; Clarimón, J; Ferrer, I; Gelpi, E; Lleó, A
Acta neuropathologica
125
201-13
2013
Mostrar Resumo
Autosomal-dominant Alzheimer disease (ADAD) is a genetic disorder caused by mutations in Amyloid Precursor Protein (APP) or Presenilin (PSEN) genes. Studies from families with ADAD have been critical to support the amyloid cascade hypothesis of Alzheimer disease (AD), the basis for the current development of amyloid-based disease-modifying therapies in sporadic AD (SAD). However, whether the pathological changes in APP processing in the CNS in ADAD are similar to those observed in SAD remains unclear. In this study, we measured β-site APP-cleaving enzyme (BACE) protein levels and activity, APP and APP C-terminal fragments in brain samples from subjects with ADAD carrying APP or PSEN1 mutations (n = 18), patients with SAD (n = 27) and age-matched controls (n = 22). We also measured sAPPβ and BACE protein levels, as well as BACE activity, in CSF from individuals carrying PSEN1 mutations (10 mutation carriers and 7 non-carrier controls), patients with SAD (n = 32) and age-matched controls (n = 11). We found that in the brain, the pattern in ADAD was characterized by an increase in APP β-C-terminal fragment (β-CTF) levels despite no changes in BACE protein levels or activity. In contrast, the pattern in SAD in the brain was mainly characterized by an increase in BACE levels and activity, with less APP β-CTF accumulation than ADAD. In the CSF, no differences were found between groups in BACE activity or expression or sAPPβ levels. Taken together, these data suggest that the physiopathological events underlying the chronic Aβ production/clearance imbalance in SAD and ADAD are different. These differences should be considered in the design of intervention trials in AD. | | 23224319
|
Mechanisms that lessen benefits of β-secretase reduction in a mouse model of Alzheimer's disease.
Devi, L; Ohno, M
Translational psychiatry
3
e284
2013
Mostrar Resumo
The β-secretase enzyme BACE1 (β-site amyloid precursor protein-cleaving enzyme 1), which initiates amyloid-β (Aβ) production, is an excellent therapeutic target for Alzheimer's disease (AD). However, recent evidence raises concern that BACE1-inhibiting approaches may encounter dramatic declines in their abilities to ameliorate AD-like pathology and memory deficits during disease progression. Here, we used BACE1 haploinsufficiency as a therapeutic relevant model to evaluate the efficacy of partial inhibition of this enzyme. Specifically, we crossed BACE1(+/-) mice with 5XFAD transgenic mice and investigated the mechanisms by which Aβ accumulation and related memory impairments become less sensitive to rescue by BACE1(+/-) reduction. Haploinsufficiency lowered BACE1 expression by ∼50% in 5XFAD mice regardless of age in concordance with reduction in gene copy number. However, profound Aβ plaque pathology and memory deficits concomitant with BACE1 equivalent to wild-type control levels remained in BACE1(+/-)·5XFAD mice with advanced age (15-18 months old). Therefore, BACE1 haploinsufficiency is not sufficient to block the elevation of BACE1 expression (approximately twofold), which is also reported to occur during human AD progression, in 5XFAD mice. Our investigation revealed that PERK (PKR-endoplasmic reticulum-related kinase)-dependent activation of eIF2α (eukaryotic translation initiation factor-2α) accounts for the persistent BACE1 upregulation in BACE1(+/-)·5XFAD mouse brains at 15-18 months of age. Moreover, BACE1 haploinsufficiency was also no longer able to prevent reduction in the expression of neprilysin, a crucial Aβ-degrading enzyme, in 5XFAD mice with advanced age. These findings demonstrate that partial BACE1 suppression cannot attenuate deleterious BACE1-elevating or neprilysin-reducing mechanisms, limiting its capabilities to reduce cerebral Aβ accumulation and rescue memory defects during the course of AD development. | Western Blotting | 23880880
|
Can platelet BACE1 levels be used as a biomarker for Alzheimer's disease? Proof-of-concept study.
Decourt, B; Walker, A; Gonzales, A; Malek-Ahmadi, M; Liesback, C; Davis, KJ; Belden, CM; Jacobson, SA; Sabbagh, MN
Platelets
24
235-8
2013
Mostrar Resumo
To date there is no validated peripheral biomarker to assist with the clinical diagnosis of Alzheimer's disease (AD). Platelet proteins have been studied as AD biomarkers with relative success. In this study, we investigated whether platelet BACE1 levels differ between AD and cognitively normal (CN) control patients. Using a newly developed ELISA method, we found that BACE1 levels were significantly lower in AD compared to CN subjects. These data were supported by the observation that several BACE1 isoforms, identified by Western blotting, were also lower in AD platelets. This proof-of-concept study provides evidence for testing platelet BACE1 levels as a peripheral AD biomarker using a novel, sensitive and inexpensive method. | | 22775589
|
BACE1 levels by APOE genotype in non-demented and Alzheimer's post-mortem brains.
Decourt, B; Gonzales, A; Beach, TG; Malek-Ahmadi, M; Walker, A; Sue, L; Walker, DG; Sabbagh, MN
Current Alzheimer research
10
309-15
2013
Mostrar Resumo
The APOE genotype is a known susceptibility factor for Alzheimer's disease (AD). It is apparent that the presence of the APOE ε40 allele increases the risk for developing AD, lowers the age of onset in AD, and may influence the pathological burden seen in AD. In this study, we asked whether BACE1 levels differ by APOE genotype in the AD and non-demented (ND) brain. We isolated mid-frontal cortex (MFC) and mid-temporal cortex (MTC) from post-mortem ND and AD subjects that were APOE ε3/3, ε3/4, ε4/4 carriers. All AD subjects met NINDS-ADRDA and NIA-Reagan criteria for a diagnosis of AD. The MFC and MTC were homogenized and the lysates underwent ELISA and Western blotting for BACE1. The ELISA revealed that total BACE1 levels were lower in the MFC of AD compared to ND subjects. Furthermore, in APOE ε4 carriers BACE1 levels were lower than ε3/3 carriers in the ND frontal cortex. No difference in BACE1 levels was observed in AD MFC and in ND and AD MTC tissues. The ELISA results were confirmed by Western blotting. Our data suggest that brain BACEl levels may be influenced by the apolipoprotein E genotype before the onset of AD, providing an alternative explanation for the lower amyloid beta 42 levels in CSF in ND and AD subjects. | | 23036023
|
7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer's disease.
Devi, L; Ohno, M
Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology
37
434-44
2012
Mostrar Resumo
Increasing evidence suggests that reductions in brain-derived neurotrophic factor (BDNF) and its receptor tyrosine receptor kinase B (TrkB) may have a role in the pathogenesis of Alzheimer's disease (AD). However, the efficacy and safety profile of BDNF therapy (eg, gene delivery) remains to be established toward clinical trials. Here, we evaluated the effects of 7,8-dihydroxyflavone (7,8-DHF), a recently identified small-molecule TrkB agonist that can pass the blood-brain barrier, in the 5XFAD transgenic mouse model of AD. 5XFAD mice at 12-15 months of age and non-transgenic littermate controls received systemic administration of 7,8-DHF (5 mg/kg, i.p.) once daily for 10 consecutive days. We found that 7,8-DHF rescued memory deficits of 5XFAD mice in the spontaneous alternation Y-maze task. 5XFAD mice showed impairments in the hippocampal BDNF-TrkB pathway, as evidenced by significant reductions in BDNF, TrkB receptors, and phosphorylated TrkB. 7,8-DHF restored deficient TrkB signaling in 5XFAD mice without affecting endogenous BDNF levels. Meanwhile, 5XFAD mice exhibited elevations in the β-secretase enzyme (BACE1) that initiates amyloid-β (Aβ) generation, as observed in sporadic AD. Interestingly, 7,8-DHF blocked BACE1 elevations and lowered levels of the β-secretase-cleaved C-terminal fragment of amyloid precursor protein (C99), Aβ40, and Aβ42 in 5XFAD mouse brains. Furthermore, BACE1 expression was decreased by 7,8-DHF in wild-type mice, suggesting that BDNF-TrkB signaling is also important for downregulating baseline levels of BACE1. Together, our findings indicate that TrkB activation with systemic 7,8-DHF can ameliorate AD-associated memory deficits, which may be, at least in part, attributable to reductions in BACE1 expression and β-amyloidogenesis. | | 21900882
|
Mitochondrial dysfunction and accumulation of the β-secretase-cleaved C-terminal fragment of APP in Alzheimer's disease transgenic mice.
Devi, L; Ohno, M
Neurobiology of disease
45
417-24
2012
Mostrar Resumo
Mitochondrial dysfunction is an early feature of Alzheimer's disease (AD) and may play an important role in the pathogenesis of disease. Emerging evidence indicates that amyloid-β (Aβ) peptides enter mitochondria and may thereby disrupt mitochondrial function in brains of AD patients and transgenic model mice. However, it remains to be determined whether the β-cleaved C-terminal fragment (C99), another neurotoxic fragment of amyloid precursor protein (APP), may accumulate in mitochondria of neurons affected by AD. Using immunoblotting, digitonin fractionation and immunofluorescence labeling techniques, we found that C99 is targeted to mitochondria, in particular, to the mitoplast (i.e., inner membrane and matrix compartments) in brains of AD transgenic mice (5XFAD model). Furthermore, full-length APP (fl-APP) was also identified in mitochondrial fractions of 5XFAD mice. Remarkably, partial deletion of the β-site APP-cleaving enzyme 1 (BACE1(+/-)) almost completely abolished mitochondrial targeting of C99 and fl-APP in 5XFAD mice at 6 months of age. However, substantial amounts of C99 and fl-APP accumulation remained in mitochondria of 12-month-old BACE1(+/-)·5XFAD mouse brains. Consistent with these changes in mitochondrial C99/fl-APP levels, BACE1(+/-) deletion age-dependently rescued mitochondrial dysfunction in 5XFAD mice, as assessed by cytochrome c release from mitochondria, reduced redox or complex activities and oxidative DNA damage. Moreover, BACE1(+/-) deletion also improved memory deficits as tested by the spontaneous alternation Y-maze task in 5XFAD mice at 6 months but not at 12 months of age. Taken together, our findings suggest that mitochondrial accumulation of C99 and fl-APP may occur through BACE1-dependent mechanisms and contribute to inducing mitochondrial dysfunction and cognitive impairments associated with AD. | | 21933711
|
Mechanisms underlying insulin deficiency-induced acceleration of β-amyloidosis in a mouse model of Alzheimer's disease.
Devi, L; Alldred, MJ; Ginsberg, SD; Ohno, M
PloS one
7
e32792
2012
Mostrar Resumo
Although evidence is accumulating that diabetes mellitus is an important risk factor for sporadic Alzheimer's disease (AD), the mechanisms by which defects in insulin signaling may lead to the acceleration of AD progression remain unclear. In this study, we applied streptozotocin (STZ) to induce experimental diabetes in AD transgenic mice (5XFAD model) and investigated how insulin deficiency affects the β-amyloidogenic processing of amyloid precursor protein (APP). Two and half months after 5XFAD mice were treated with STZ (90 mg/kg, i.p., once daily for two consecutive days), they showed significant reductions in brain insulin levels without changes in insulin receptor expression. Concentrations of cerebral amyloid-β peptides (Aβ40 and Aβ42) were significantly increased in STZ-treated 5XFAD mice as compared with vehicle-treated 5XFAD controls. Importantly, STZ-induced insulin deficiency upregulated levels of both β-site APP cleaving enzyme 1 (BACE1) and full-length APP in 5XFAD mouse brains, which was accompanied by dramatic elevations in the β-cleaved C-terminal fragment (C99). Interestingly, BACE1 mRNA levels were not affected, whereas phosphorylation of the translation initiation factor eIF2α, a mechanism proposed to mediate the post-transcriptional upregulation of BACE1, was significantly elevated in STZ-treated 5XFAD mice. Meanwhile, levels of GGA3, an adapter protein responsible for sorting BACE1 to lysosomal degradation, are indistinguishable between STZ- and vehicle-treated 5XFAD mice. Moreover, STZ treatments did not affect levels of Aβ-degrading enzymes such as neprilysin and insulin-degrading enzyme (IDE) in 5XFAD brains. Taken together, our findings provide a mechanistic foundation for a link between diabetes and AD by demonstrating that insulin deficiency may change APP processing to favor β-amyloidogenesis via the translational upregulation of BACE1 in combination with elevations in its substrate, APP. | | 22403710
|
Oxidative lipid modification of nicastrin enhances amyloidogenic γ-secretase activity in Alzheimer's disease.
A-Ryeong Gwon,Jong-Sung Park,Thiruma V Arumugam,Yong-Kook Kwon,Sic L Chan,Seol-Hee Kim,Sang-Ha Baik,Sunghee Yang,Young-Kwang Yun,Yuri Choi,Saerom Kim,Sung-Chun Tang,Dong-Hoon Hyun,Aiwu Cheng,Charles E Dann,Michel Bernier,Jaewon Lee,William R Markesbery,Mark P Mattson,Dong-Gyu Jo
Aging cell
11
2012
Mostrar Resumo
The cause of elevated level of amyloid β-peptide (Aβ42) in common late-onset sporadic [Alzheimer's disease (AD)] has not been established. Here, we show that the membrane lipid peroxidation product 4-hydroxynonenal (HNE) is associated with amyloid and neurodegenerative pathologies in AD and that it enhances γ-secretase activity and Aβ42 production in neurons. The γ-secretase substrate receptor, nicastrin, was found to be modified by HNE in cultured neurons and in brain specimens from patients with AD, in which HNE-nicastrin levels were found to be correlated with increased γ-secretase activity and Aβ plaque burden. Furthermore, HNE modification of nicastrin enhanced its binding to the γ-secretase substrate, amyloid precursor protein (APP) C99. In addition, the stimulation of γ-secretase activity and Aβ42 production by HNE were blocked by an HNE-scavenging histidine analog in a 3xTgAD mouse model of AD. These findings suggest a specific molecular mechanism by which oxidative stress increases Aβ42 production in AD and identify HNE as a novel therapeutic target upstream of the γ-secretase cleavage of APP. | | 22404891
|