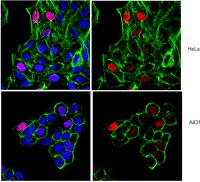
The Drosophila melanogaster CHD1 chromatin remodeling factor modulates global chromosome structure and counteracts HP1a and H3K9me2.
Bugga, L; McDaniel, IE; Engie, L; Armstrong, JA
PloS one
8
e59496
2013
Afficher le résumé
CHD1 is a conserved chromatin remodeling factor that localizes to active genes and functions in nucleosome assembly and positioning as well as histone turnover. Mouse CHD1 is required for the maintenance of stem cell pluripotency while human CHD1 may function as a tumor suppressor. To investigate the action of CHD1 on higher order chromatin structure in differentiated cells, we examined the consequences of loss of CHD1 and over-expression of CHD1 on polytene chromosomes from salivary glands of third instar Drosophila melanogaster larvae. We observed that chromosome structure is sensitive to the amount of this remodeler. Loss of CHD1 resulted in alterations of chromosome structure and an increase in the heterochromatin protein HP1a, while over-expression of CHD1 disrupted higher order chromatin structure and caused a decrease in levels of HP1a. Over-expression of an ATPase inactive form of CHD1 did not result in severe chromosomal defects, suggesting that the ATPase activity is required for this in vivo phenotype. Interestingly, changes in CHD1 protein levels did not correlate with changes in the levels of the euchromatin mark H3K4me3 or elongating RNA Polymerase II. Thus, while CHD1 is localized to transcriptionally active regions of the genome, it can function to alter the levels of HP1a, perhaps through changes in methylation of H3K9. | Immunofluorescence | 23533627
 |
A dual role for SAGA-associated factor 29 (SGF29) in ER stress survival by coordination of both histone H3 acetylation and histone H3 lysine-4 trimethylation.
Schram, AW; Baas, R; Jansen, PW; Riss, A; Tora, L; Vermeulen, M; Timmers, HT
PloS one
8
e70035
2013
Afficher le résumé
The SGF29 protein binds to tri-methylated lysine-4 of histone H3 (H3K4me3), which is a histone modification associated with active promoters. Human SGF29 is a subunit of the histone acetyltransferase module of the SAGA (Spt-Ada-Gcn5 acetyltransferase) and ATAC (Ada-Two-A-containing 2A) co-activator complexes. Previous work revealed that the SAGA complex is recruited to endoplasmic reticulum (ER) stress target genes and required for their induction. Here, we report the involvement of SGF29 in the survival of human cells from ER stress. SGF29 knockdown results in impaired transcription of the ER stress genes GRP78 and CHOP. Besides histone H3K14 acetylation, we find that SGF29 is also required for the maintenance of H3K4me3 at these genes, which is already present prior to ER stress. Reduced levels of H3K4me3 in the absence of SGF29 correlate with a decreased association of ASH2L, which is a core component of the SET1/MLL complexes, to GFP78 and CHOP. In conclusion, our results suggest that the H3K4me3-binding protein SGF29 plays a central and dual role in the ER stress response. Prior to ER stress, the protein coordinates H3K4me3 levels, thereby maintaining a 'poised' chromatin state on ER stress target gene promoters. Following ER stress induction, SGF29 is required for increased H3K14 acetylation on these genes, which then results in full transcriptional activation, thereby promoting cell survival. | | 23894581
|
Chromatin signatures at transcriptional start sites separate two equally populated yet distinct classes of intergenic long noncoding RNAs.
Marques, AC; Hughes, J; Graham, B; Kowalczyk, MS; Higgs, DR; Ponting, CP
Genome biology
14
R131
2013
Afficher le résumé
Mammalian transcriptomes contain thousands of long noncoding RNAs (lncRNAs). Some lncRNAs originate from intragenic enhancers which, when active, behave as alternative promoters producing transcripts that are processed using the canonical signals of their host gene. We have followed up this observation by analyzing intergenic lncRNAs to determine the extent to which they might also originate from intergenic enhancers.We integrated high-resolution maps of transcriptional initiation and transcription to annotate a conservative set of intergenic lncRNAs expressed in mouse erythroblasts. We subclassified intergenic lncRNAs according to chromatin status at transcriptional initiation regions, defined by relative levels of histone H3K4 mono- and trimethylation. These transcripts are almost evenly divided between those arising from enhancer-associated (elncRNA) or promoter-associated (plncRNA) elements. These two classes of 5' capped and polyadenylated RNA transcripts are indistinguishable with regard to their length, number of exons or transcriptional orientation relative to their closest neighboring gene. Nevertheless, elncRNAs are more tissue-restricted, less highly expressed and less well conserved during evolution. Of considerable interest, we found that expression of elncRNAs, but not plncRNAs, is associated with enhanced expression of neighboring protein-coding genes during erythropoiesis.We have determined globally the sites of initiation of intergenic lncRNAs in erythroid cells, allowing us to distinguish two similarly abundant classes of transcripts. Different correlations between the levels of elncRNAs, plncRNAs and expression of neighboring genes suggest that functional lncRNAs from the two classes may play contrasting roles in regulating the transcript abundance of local or distal loci. | | 24289259
|
An efficient immunodetection method for histone modifications in plants.
Nic-Can, G; Hernández-Castellano, S; Kú-González, A; Loyola-Vargas, VM; De-la-Peña, C
Plant methods
9
47
2013
Afficher le résumé
Epigenetic mechanisms can be highly dynamic, but the cross-talk among them and with the genome is still poorly understood. Many of these mechanisms work at different places in the cell and at different times of organism development. Covalent histone modifications are one of the most complex and studied epigenetic mechanisms involved in cellular reprogramming and development in plants. Therefore, the knowledge of the spatial distribution of histone methylation in different tissues is important to understand their behavior on specific cells.Based on the importance of epigenetic marks for biology, we present a simplified, inexpensive and efficient protocol for in situ immunolocalization on different tissues such as flowers, buds, callus, somatic embryo and meristematic tissue from several plants of agronomical and biological importance. Here, we fully describe all the steps to perform the localization of histone modifications. Using this method, we were able to visualize the distribution of H3K4me3 and H3K9me2 without loss of histological integrity of tissues from several plants, including Agave tequilana, Capsicum chinense, Coffea canephora and Cedrela odorata, as well as Arabidopsis thaliana.There are many protocols to study chromatin modifications; however, most of them are expensive, difficult and require sophisticated equipment. Here, we provide an efficient protocol for in situ localization of histone methylation that dispenses with the use of expensive and sensitive enzymes. The present method can be used to investigate the cellular distribution and localization of a wide array of proteins, which could help to clarify the biological role that they play at specific times and places in different tissues of various plant species. | | 24341414
|
Coordinate H3K9 and DNA methylation silencing of ZNFs in toxicant-induced malignant transformation.
Severson, PL; Tokar, EJ; Vrba, L; Waalkes, MP; Futscher, BW
Epigenetics
8
1080-8
2013
Afficher le résumé
Genome-wide disruption of the epigenetic code is a hallmark of malignancy that encompasses many distinct, highly interactive modifications. Delineating the aberrant epigenome produced during toxicant-mediated malignant transformation will help identify the underlying epigenetic drivers of environmental toxicant-induced carcinogenesis. Gene promoter DNA methylation and gene expression profiling of arsenite-transformed prostate epithelial cells showed a negative correlation between gene expression changes and DNA methylation changes; however, less than 10% of the genes with increased promoter methylation were downregulated. Studies described herein confirm that a majority of the DNA hypermethylation events occur at H3K27me3 marked genes that were already transcriptionally repressed. In contrast to aberrant DNA methylation targeting H3K27me3 pre-marked silent genes, we found that actively expressed C2H2 zinc finger genes (ZNFs) marked with H3K9me3 on their 3' ends, were the favored targets of DNA methylation linked gene silencing. DNA methylation coupled, H3K9me3 mediated gene silencing of ZNF genes was widespread, occurring at individual ZNF genes on multiple chromosomes and across ZNF gene family clusters. At ZNF gene promoters, H3K9me3 and DNA hypermethylation replaced H3K4me3, resulting in a widespread downregulation of ZNF gene expression, which accounted for 8% of all the downregulated genes in the arsenical-transformed cells. In summary, these studies associate toxicant exposure with widespread silencing of ZNF genes by DNA hypermethylation-linked H3K9me3 spreading, further implicating epigenetic dysfunction as a driver of toxicant associated carcinogenesis. | | 23974009
|
Dynamics and memory of heterochromatin in living cells.
Hathaway, NA; Bell, O; Hodges, C; Miller, EL; Neel, DS; Crabtree, GR
Cell
149
1447-60
2011
Afficher le résumé
Posttranslational histone modifications are important for gene regulation, yet the mode of propagation and the contribution to heritable gene expression states remains controversial. To address these questions, we developed a chromatin in vivo assay (CiA) system employing chemically induced proximity to initiate and terminate chromatin modifications in living cells. We selectively recruited HP1α to induce H3K9me3-dependent gene silencing and describe the kinetics and extent of chromatin modifications at the Oct4 locus in fibroblasts and pluripotent cells. H3K9me3 propagated symmetrically and continuously at average rates of ~0.18 nucleosomes/hr to produce domains of up to 10 kb. After removal of the HP1α stimulus, heterochromatic domains were heritably transmitted, undiminished through multiple cell generations. Our data enabled quantitative modeling of reaction kinetics, which revealed that dynamic competition between histone marking and turnover, determines the boundaries and stability of H3K9me3 domains. This framework predicts the steady-state dynamics and spatial features of the majority of euchromatic H3K9me3 domains over the genome. | | 22704655
|
A map of the cis-regulatory sequences in the mouse genome.
Shen, Yin, et al.
Nature, 488: 116-20 (2012)
2011
Afficher le résumé
The laboratory mouse is the most widely used mammalian model organism in biomedical research. The 2.6 × 10(9) bases of the mouse genome possess a high degree of conservation with the human genome, so a thorough annotation of the mouse genome will be of significant value to understanding the function of the human genome. So far, most of the functional sequences in the mouse genome have yet to be found, and the cis-regulatory sequences in particular are still poorly annotated. Comparative genomics has been a powerful tool for the discovery of these sequences, but on its own it cannot resolve their temporal and spatial functions. Recently, ChIP-Seq has been developed to identify cis-regulatory elements in the genomes of several organisms including humans, Drosophila melanogaster and Caenorhabditis elegans. Here we apply the same experimental approach to a diverse set of 19 tissues and cell types in the mouse to produce a map of nearly 300,000 murine cis-regulatory sequences. The annotated sequences add up to 11% of the mouse genome, and include more than 70% of conserved non-coding sequences. We define tissue-specific enhancers and identify potential transcription factors regulating gene expression in each tissue or cell type. Finally, we show that much of the mouse genome is organized into domains of coordinately regulated enhancers and promoters. Our results provide a resource for the annotation of functional elements in the mammalian genome and for the study of mechanisms regulating tissue-specific gene expression. | | 22763441
|
Polycomb eviction as a new distant enhancer function.
Vernimmen, D; Lynch, MD; De Gobbi, M; Garrick, D; Sharpe, JA; Sloane-Stanley, JA; Smith, AJ; Higgs, DR
Genes & development
25
1583-8
2010
Afficher le résumé
Remote distal enhancers may be located tens or thousands of kilobases away from their promoters. How they control gene expression is still poorly understood. Here, we analyze the influence of a remote enhancer on the balance between repression (Polycomb-PcG) and activation (Trithorax-TrxG) of a developmentally regulated gene associated with a CpG island. We reveal its essential, nonredundant role in clearing the PcG complex and H3K27me3 from the CpG island. In the absence of the enhancer, the H3K27me3 demethylase (JMJD3) is not recruited to the CpG island. We propose a new role of long-range regulatory elements in removing repressive PcG complexes. | | 21828268
|
Histone crosstalk directed by H2B ubiquitination is required for chromatin boundary integrity.
Ma, MK; Heath, C; Hair, A; West, AG
PLoS genetics
7
e1002175
2010
Afficher le résumé
Genomic maps of chromatin modifications have provided evidence for the partitioning of genomes into domains of distinct chromatin states, which assist coordinated gene regulation. The maintenance of chromatin domain integrity can require the setting of boundaries. The HS4 insulator element marks the 3' boundary of a heterochromatin region located upstream of the chicken β-globin gene cluster. Here we show that HS4 recruits the E3 ligase RNF20/BRE1A to mediate H2B mono-ubiquitination (H2Bub1) at this insulator. Knockdown experiments show that RNF20 is required for H2Bub1 and processive H3K4 methylation. Depletion of RNF20 results in a collapse of the active histone modification signature at the HS4 chromatin boundary, where H2Bub1, H3K4 methylation, and hyperacetylation of H3, H4, and H2A.Z are rapidly lost. A remarkably similar set of events occurs at the HSA/HSB regulatory elements of the FOLR1 gene, which mark the 5' boundary of the same heterochromatin region. We find that persistent H2Bub1 at the HSA/HSB and HS4 elements is required for chromatin boundary integrity. The loss of boundary function leads to the sequential spreading of H3K9me2, H3K9me3, and H4K20me3 over the entire 50 kb FOLR1 and β-globin region and silencing of FOLR1 expression. These findings show that the HSA/HSB and HS4 boundary elements direct a cascade of active histone modifications that defend the FOLR1 and β-globin gene loci from the pervasive encroachment of an adjacent heterochromatin domain. We propose that many gene loci employ H2Bub1-dependent boundaries to prevent heterochromatin spreading. | | 21811414
|
Arabidopsis COMPASS-like complexes mediate histone H3 lysine-4 trimethylation to control floral transition and plant development.
Jiang, D; Kong, NC; Gu, X; Li, Z; He, Y
PLoS genetics
7
e1001330
2010
Afficher le résumé
Histone H3 lysine-4 (H3K4) methylation is associated with transcribed genes in eukaryotes. In Drosophila and mammals, both di- and tri-methylation of H3K4 are associated with gene activation. In contrast to animals, in Arabidopsis H3K4 trimethylation, but not mono- or di-methylation of H3K4, has been implicated in transcriptional activation. H3K4 methylation is catalyzed by the H3K4 methyltransferase complexes known as COMPASS or COMPASS-like in yeast and mammals. Here, we report that Arabidopsis homologs of the COMPASS and COMPASS-like complex core components known as Ash2, RbBP5, and WDR5 in humans form a nuclear subcomplex during vegetative and reproductive development, which can associate with multiple putative H3K4 methyltransferases. Loss of function of ARABIDOPSIS Ash2 RELATIVE (ASH2R) causes a great decrease in genome-wide H3K4 trimethylation, but not in di- or mono-methylation. Knockdown of ASH2R or the RbBP5 homolog suppresses the expression of a crucial Arabidopsis floral repressor, FLOWERING LOCUS C (FLC), and FLC homologs resulting in accelerated floral transition. ASH2R binds to the chromatin of FLC and FLC homologs in vivo and is required for H3K4 trimethylation, but not for H3K4 dimethylation in these loci; overexpression of ASH2R causes elevated H3K4 trimethylation, but not H3K4 dimethylation, in its target genes FLC and FLC homologs, resulting in activation of these gene expression and consequent late flowering. These results strongly suggest that H3K4 trimethylation in FLC and its homologs can activate their expression, providing concrete evidence that H3K4 trimethylation accumulation can activate eukaryotic gene expression. Furthermore, our findings suggest that there are multiple COMPASS-like complexes in Arabidopsis and that these complexes deposit trimethyl but not di- or mono-methyl H3K4 in target genes to promote their expression, providing a molecular explanation for the observed coupling of H3K4 trimethylation (but not H3K4 dimethylation) with active gene expression in Arabidopsis. | Western Blotting | 21423667
|