Combining the differentiating effect of panobinostat with the apoptotic effect of arsenic trioxide leads to significant survival benefit in a model of t(8;21) acute myeloid leukemia.
Salmon, JM; Bots, M; Vidacs, E; Stanley, KL; Atadja, P; Zuber, J; Johnstone, RW
Clinical epigenetics
7
2
2015
Afficher le résumé
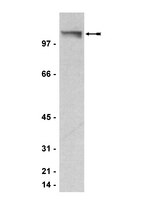
One of the most frequently found abnormalities in acute myeloid leukemia (AML) is the t(8;21)(q22;q22) translocation, which is seen in around 15% of patients. This translocation results in the production of the AML1/ETO (A/E) fusion protein and commonly involves cooperating activating mutations of RAS. AE9a encodes a C-terminally truncated A/E protein of 575 amino acids that retains the ability to recruit histone deacetylases (HDACs). Expression of AE9a leads to rapid development of leukemia in experimental mouse systems. We have recently shown that treatment of mice bearing A/E9a;Nras (G12D) tumors with the histone deacetylase inhibitor (HDACi) panobinostat leads to degradation of the A/E9a fusion protein, cell cycle arrest, differentiation of AML blasts into mature granulocytes and prolonged survival. Herein, we sought to enhance this therapeutic effect.Combined treatment of mice bearing A/E9a;Nras (G12D) leukemias with panobinostat and arsenic trioxide (ATO) resulted in a significant survival advantage compared to mice treated with either agent alone. Moreover, some of the mice treated with the panobinostat/ATO combination showed complete tumor responses and remained in remission for over 220 days. Panobinostat caused differentiation of A/E9a;Nras (G12D) cells while ATO induced apoptosis of the leukemic cells, an effect that was enhanced following co-treatment with panobinostat.Our results indicate that leukemic blast differentiation mediated by panobinostat combined with induction of apoptosis by ATO could be therapeutically beneficial and should be considered for patients with t(8;21) AML. | Western Blotting | 25628765
 |
Senescence induced by RECQL4 dysfunction contributes to Rothmund-Thomson syndrome features in mice.
Lu, H; Fang, EF; Sykora, P; Kulikowicz, T; Zhang, Y; Becker, KG; Croteau, DL; Bohr, VA
Cell death & disease
5
e1226
2014
Afficher le résumé
Cellular senescence refers to irreversible growth arrest of primary eukaryotic cells, a process thought to contribute to aging-related degeneration and disease. Deficiency of RecQ helicase RECQL4 leads to Rothmund-Thomson syndrome (RTS), and we have investigated whether senescence is involved using cellular approaches and a mouse model. We first systematically investigated whether depletion of RECQL4 and the other four human RecQ helicases, BLM, WRN, RECQL1 and RECQL5, impacts the proliferative potential of human primary fibroblasts. BLM-, WRN- and RECQL4-depleted cells display increased staining of senescence-associated β-galactosidase (SA-β-gal), higher expression of p16(INK4a) or/and p21(WAF1) and accumulated persistent DNA damage foci. These features were less frequent in RECQL1- and RECQL5-depleted cells. We have mapped the region in RECQL4 that prevents cellular senescence to its N-terminal region and helicase domain. We further investigated senescence features in an RTS mouse model, Recql4-deficient mice (Recql4(HD)). Tail fibroblasts from Recql4(HD) showed increased SA-β-gal staining and increased DNA damage foci. We also identified sparser tail hair and fewer blood cells in Recql4(HD) mice accompanied with increased senescence in tail hair follicles and in bone marrow cells. In conclusion, dysfunction of RECQL4 increases DNA damage and triggers premature senescence in both human and mouse cells, which may contribute to symptoms in RTS patients. | Immunofluorescence | 24832598
|
SIRT1 stabilizes PML promoting its sumoylation.
Campagna, M; Herranz, D; Garcia, MA; Marcos-Villar, L; González-Santamaría, J; Gallego, P; Gutierrez, S; Collado, M; Serrano, M; Esteban, M; Rivas, C
Cell death and differentiation
18
72-9
2010
Afficher le résumé
SIRT1, the closest mammalian homolog of yeast Sir2, is an NAD(+)-dependent deacetylase with relevant functions in cancer, aging, and metabolism among other processes. SIRT1 has a diffuse nuclear localization but is recruited to the PML nuclear bodies (PML-NBs) after PML upregulation. However, the functions of SIRT1 in the PML-NBs are unknown. In this study we show that primary mouse embryo fibroblasts lacking SIRT1 contain reduced PML protein levels that are increased after reintroduction of SIRT1. In addition, overexpression of SIRT1 in HEK-293 cells increases the amount of PML protein whereas knockdown of SIRT1 reduces the size and number of PML-NBs and the levels of PML protein in HeLa cells. SIRT1 stimulates PML sumoylation in vitro and in vivo in a deacetylase-independent manner. Importantly, the absence of SIRT1 reduces the apoptotic response of vesicular stomatitis virus-infected cells and favors the extent of this PML-sensitive virus replication. These results show a novel function of SIRT1 in the control of PML and PML-NBs. | Immunofluorescence | 20577263
|
Senescent mouse cells fail to overtly regulate the HIRA histone chaperone and do not form robust Senescence Associated Heterochromatin Foci.
Alyssa L Kennedy,Tony McBryan,Greg H Enders,F Brad Johnson,Rugang Zhang,Peter D Adams
Cell division
5
2009
Afficher le résumé
Cellular senescence is a permanent growth arrest that occurs in response to cellular stressors, such as telomere shortening or activation of oncogenes. Although the process of senescence growth arrest is somewhat conserved between mouse and human cells, there are some critical differences in the molecular pathways of senescence between these two species. Recent studies in human fibroblasts have defined a cell signaling pathway that is initiated by repression of a specific Wnt ligand, Wnt2. This, in turn, activates a histone chaperone HIRA, and culminates in formation of specialized punctate domains of facultative heterochromatin, called Senescence-Associated Heterochromatin Foci (SAHF), that are enriched in the histone variant, macroH2A. SAHF are thought to repress expression of proliferation-promoting genes, thereby contributing to senescence-associated proliferation arrest. We asked whether this Wnt2-HIRA-SAHF pathway is conserved in mouse fibroblasts. Article en texte intégral | | 20569479
|
Assembly dynamics of PML nuclear bodies in living cells.
Brand, P; Lenser, T; Hemmerich, P
PMC biophysics
3
3
2009
Afficher le résumé
The mammalian cell nucleus contains a variety of organelles or nuclear bodies which contribute to key nuclear functions. Promyelocytic leukemia nuclear bodies (PML NBs) are involved in the regulation of apoptosis, antiviral responses, the DNA damage response and chromatin structure, but their precise biochemical function in these nuclear pathways is unknown. One strategy to tackle this problem is to assess the biophysical properties of the component parts of these macromolecular assemblies in living cells. In this study we determined PML NB assembly dynamics by live cell imaging, combined with mathematical modeling. For the first time, dynamics of PML body formation were measured in cells lacking endogenous PML. We show that all six human nuclear PML isoforms are able to form nuclear bodies in PML negative cells. All isoforms exhibit individual exchange rates at NBs in PML positive cells but PML I, II, III and IV are static at nuclear bodies in PML negative cells, suggesting that these isoforms require additional protein partners for efficient exchange. PML V turns over at PML Nbs very slowly supporting the idea of a structural function for this isoform. We also demonstrate that SUMOylation of PML at Lysine positions K160 and/or K490 are required for nuclear body formation in vivo.We propose a model in which the isoform specific residence times of PML provide both, structural stability to function as a scaffold and flexibility to attract specific nuclear proteins for efficient biochemical reactions at the surface of nuclear bodies.MCS code: 92C37. Article en texte intégral | | 20205709
|
HDAC3 as a molecular chaperone for shuttling phosphorylated TR2 to PML: a novel deacetylase activity-independent function of HDAC3.
Gupta, P; Ho, PC; Ha, SG; Lin, YW; Wei, LN
PloS one
4
e4363
2009
Afficher le résumé
TR2 is an orphan nuclear receptor specifically expressed in early embryos (Wei and Hsu, 1994), and a transcription factor for transcriptional regulation of important genes in stem cells including the gate keeper Oct4 (Park et al. 2007). TR2 is known to function as an activator (Wei et al. 2000), or a repressor (Chinpaisal et al., 1998, Gupta et al. 2007). Due to the lack of specific ligands, mechanisms triggering its activator or repressor function have remained puzzling for decades. Recently, we found that all-trans retinoic acid (atRA) triggers the activation of extracellular-signal-regulated kinase 2 (ERK2), which phosphorylates TR2 and stimulates its partitioning to promyelocytic leukemia (PML) nuclear bodies, thereby converting the activator function of TR2 into repression (Gupta et al. 2008; Park et al. 2007). Recruitment of TR2 to PML is a crucial step in the conversion of TR2 from an activator to a repressor. However, it is unclear how phosphorylated TR2 is recruited to PML, an essential step in converting TR2 from an activator to a repressor. In the present study, we use both in vitro and in vivo systems to address the problem of recruiting TR2 to PML nuclear bodies. First, we identify histone deacetylase 3 (HDAC3) as an effector molecule. HDAC3 is known to interact with TR2 (Franco et al. 2001) and this interaction is enhanced by the atRA-stimulated phosphorylation of TR2 at Thr-210 (Gupta et al. 2008). Secondly, in this study, we also find that the carrier function of HDAC3 is independent of its deacetylase activity. Thirdly, we find another novel activity of atRA that stimulates nuclear enrichment of HDAC3 to form nuclear complex with PML, which is ERK2 independent. This is the first report identifying a deacetylase-independent function for HDAC3, which serves as a specific carrier molecule that targets a specifically phosphorylated protein to PML NBs. This is also the first study delineating how protein recruitment to PML nuclear bodies occurs, which can be stimulated by atRA in an ERK2-independent manner. These findings could provide new insights into the development of potential therapeutics and in understanding how orphan nuclear receptor activities can be regulated without ligands. Article en texte intégral | | 19204783
|
ERG-associated protein with SET domain (ESET)-Oct4 interaction regulates pluripotency and represses the trophectoderm lineage.
Yeap, LS; Hayashi, K; Surani, MA
Epigenetics & chromatin
2
12
2009
Afficher le résumé
Pluripotency, the capacity for indefinite self-renewal and differentiation into diverse cell types is a unique state exhibited by embryonic stem (ES) cells. Transcriptional regulators, such as Oct4, are critical for pluripotency, but the role of epigenetic modifiers remains to be fully elucidated.Here, we show that ERG-associated protein with SET domain (ESET), a histone methyltransferase enzyme, maintains pluripotency through repression of Cdx2, a key trophectoderm determinant, by histone H3 lysine 9 trimethylation (H3K9me3) of the promoter region. Notably, this repression is mediated through the synergistic function of small ubiquitin-related modifier (SUMO)ylated ESET and Oct4. ESET localises to the promyelocytic leukaemia (PML) nuclear bodies and is SUMOylated in ES cells. Interaction of ESET with Oct4 depends on a SUMO-interacting motif (SIM) in Oct4, which is critical for the repression of Cdx2.Loss of ESET or Oct4 results in strikingly similar phenotypes both in ES cells with their differentiation into trophectoderm cells, and in early embryos where there is a failure of development of the pluripotent inner cell mass (ICM) of blastocysts. We propose that SUMOylated ESET-Oct4 complex is critical for both the initiation and maintenance of pluripotency through repression of differentiation, particularly of the trophectoderm lineage by epigenetic silencing of Cdx2. | Western Blotting | 19811652
|
Retinoic acid-stimulated sequential phosphorylation, PML recruitment, and SUMOylation of nuclear receptor TR2 to suppress Oct4 expression.
Gupta, P; Ho, PC; Huq, MM; Ha, SG; Park, SW; Khan, AA; Tsai, NP; Wei, LN
Proceedings of the National Academy of Sciences of the United States of America
105
11424-9
2008
Afficher le résumé
We previously reported an intricate mechanism underlying the homeostasis of Oct4 expression in normally proliferating stem cell culture of P19, mediated by SUMOylation of orphan nuclear receptor TR2. In the present study, we identify a signaling pathway initiated from the nongenomic activity of all-trans retinoic acid (atRA) to stimulate complex formation of extracellular signal-regulated kinase 2 (ERK2) with its upstream kinase, mitogen-activated protein kinase kinase (MEK). The activated ERK2 phosphorylates threonine-210 (Thr-210) of TR2, stimulating its subsequent SUMOylation. Dephosphorylated TR2 recruits coactivator PCAF and functions as an activator for its target gene Oct4. Upon phosphorylation at Thr-210, TR2 increasingly associates with promyelocytic leukemia (PML) nuclear bodies, becomes SUMOylated, and recruits corepressor RIP140 to act as a repressor for its target, Oct4. To normally proliferating P19 stem cell culture, exposure to a physiological concentration of atRA triggers a rapid nongenomic signaling cascade to suppress Oct4 gene and regulate cell proliferation. Article en texte intégral | | 18682553
|
SUMOylation of Tr2 orphan receptor involves Pml and fine-tunes Oct4 expression in stem cells.
Sung Wook Park, Xinli Hu, Pawan Gupta, Ya-Ping Lin, Sung Gil Ha, Li-Na Wei
Nature structural molecular biology
14
68-75
2007
Afficher le résumé
The Tr2 orphan nuclear receptor can be SUMOylated, resulting in the replacement of coregulators recruited to the regulatory region of its endogenous target gene, Oct4. UnSUMOylated Tr2 activates Oct4, enhancing embryonal carcinoma-cell proliferation, and is localized to the promyelocytic leukemia (Pml) nuclear bodies. When its abundance is elevated, Tr2 is SUMOylated at Lys238 and seems to be released from the nuclear bodies to act as a repressor. SUMOylation of Tr2 induces an exchange of its coregulators: corepressor Rip140 replaces coactivator Pcaf, which switches Tr2 from an activator to a repressor. This involves dynamic partitioning of Tr2 into Pml-containing and Pml-free pools. These results support a model where SUMOylation-dependent partitioning and differential coregulator recruitment contribute to the maintenance of a homeostatic supply of activating, as opposed to repressive, Tr2, thus fine-tuning Oct4 expression and regulating stem-cell proliferation. | | 17187077
|
SUMO-1-dependent allosteric regulation of thymine DNA glycosylase alters subnuclear localization and CBP/p300 recruitment.
Mohan, RD; Rao, A; Gagliardi, J; Tini, M
Molecular and cellular biology
27
229-43
2007
Afficher le résumé
Previous studies have demonstrated that the base excision repair enzyme thymine DNA glycosylase (TDG) mediates recruitment of histone acetyltransferases CREB-binding protein (CBP) and p300 to DNA, suggesting a plausible role for these factors in TDG-mediated repair. Furthermore, TDG was found to potentiate CBP/p300-dependent transcription and serve as a substrate for CBP/p300 acetylation. Here, we show that the small ubiquitin-like modifier 1 (SUMO-1) protein binding activity of TDG is essential for activation of CBP and localization to promyelocytic leukemia protein oncogenic domains (PODs). SUMO-1 binding is mediated by two distinct amino- and carboxy-terminal motifs (residues 144 to 148 and 319 to 322) that are negatively regulated by DNA binding via an amino-terminal hydrophilic region (residues 1 to 121). TDG is also posttranslationally modified by covalent conjugation of SUMO-1 (sumoylation) to lysine 341. Interestingly, we found that sumoylation of TDG blocks interaction with CBP and prevents TDG acetylation in vitro. Furthermore, sumoylation effectively abrogates intermolecular SUMO-1 binding and a sumoylation-deficient mutant accumulates in PODs, suggesting that sumoylation negatively regulates translocation to these nuclear structures. These findings suggest that TDG sumoylation promotes intramolecular interactions with amino- and carboxy-terminal SUMO-1 binding motifs that dramatically alter the biochemical properties and subcellular localization of TDG. | | 17060459
|