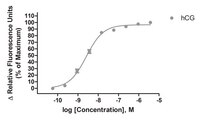
A signaling-selective, nanomolar potent allosteric low molecular weight agonist for the human luteinizing hormone receptor.
van Koppen, Chris J, et al.
Naunyn Schmiedebergs Arch. Pharmacol., 378: 503-14 (2008)
2008
Show Abstract
Luteinizing hormone (LH) and human chorionic gonadotropin (hCG) activate the LH receptor/cyclic AMP (cAMP) signaling pathway to induce ovulation. As an alternative to parenterally administered hCG to treat anovulatory infertility, orally active low molecular weight (LMW) LHR agonists have been developed at Organon. In this paper, we present the mechanism of action of a prototypic, nanomolar potent and almost full LHR agonist, Org 43553. Org 43553 interacts with the endodomain of the LHR, whereas LH acts via the N-terminal exodomain. LH stimulates the cAMP pathway with an EC50 of 35 pM, but this stimulation is not antagonized by simultaneous incubation with Org 43553. At nanomolar concentrations, LH also stimulates phospholipase C (PLC), but Org 43553 is hardly able to do so. In contrast, Org 43553 inhibits LH-induced PLC (IC50 approximately 10 nM). While Org 43553 stimulates dissociation of [125I]hCG from the LHR and reduces [125I]hCG binding, LH reduces specific [3H]Org 43553 binding. We conclude that Org 43553 is a signaling-selective, allosteric LHR agonist. We hypothesize that Org 43553 and LH induce a similar LHR conformation necessary for activating adenylyl cyclase, which initiates most, if not all, physiological responses of LH. | 18551279
 |
Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function.
Themmen APN, and Huhtaniemi, I T
Endocr. Rev., 21: 551-83 (2000)
2000
Show Abstract
The recent unraveling of structures of genes for the gonadotropin subunits and gonadotropin receptors has provided reproductive endocrinologists with new tools to study normal and pathological functions of the hypothalamic-pituitary-gonadal axis. Rare inactivating mutations that produce distinctive phenotypes of isolated LH or FSH deficiency have been discovered in gonadotropin subunit genes. In addition, there is a common polymorphism in the LHbeta subunit gene with possible clinical significance as a contributing factor to pathologies of LH-dependent gonadal functions. Both activating and inactivating mutations have been detected in the gonadotropin receptor genes, a larger number in the LH receptor gene, but so far only a few in the gene for the FSH receptor. These mutations corroborate and extend our knowledge of clinical consequences of gonadotropin resistance and inappropriate gonadotropin action. The information obtained from human mutations has been complemented by animal models with disrupted or inappropriately activated gonadotropin ligand or receptor genes. These clinical and experimental genetic disease models form a powerful tool for exploring the physiology and pathophysiology of gonadotropin function and provide an excellent example of the power of molecular biological approaches in the study of pathogenesis of diseases. | 11041448
|
Expression of human luteinizing hormone (LH) receptor: interaction with LH and chorionic gonadotropin from human but not equine, rat, and ovine species.
Jia, X C, et al.
Mol. Endocrinol., 5: 759-68 (1991)
1991
Show Abstract
Studies on human LH receptors are difficult due to the limited availability of clinical samples. Recent cloning of rat and porcine LH receptor cDNAs indicated that these binding sites are single polypeptides of the G-protein-coupled receptor family with seven transmembrane domains. Based on the conserved sequences of rat and porcine receptors, we performed reverse transcription polymerase chain reaction, using human ovarian mRNA as template and obtained partial human LH receptor cDNA clones. Further screening of a human ovary cDNA library and subsequent ligation of individual cDNA clones generated a human LH receptor cDNA containing the entire amino acid-coding region. Sequence analysis indicated that the human receptor cDNA displays 89% and 82% homology at the nucleotide level with its porcine and rat counterparts, respectively. A region spanning the second extracellular and third transmembrane domains is highly conserved among the human LH, FSH, and TSH receptors. The ovarian LH receptor clone is, however, significantly different from an incompletely spliced LH receptor cDNA recently obtained from a human thyroid library. Unlike the thyroid clone, the ovarian LH receptor cDNA could be expressed in the human fetal kidney cell line (293), and radioligand receptor assay identified high affinity (Kd, 1.2 x 10(-10) M) LH/hCG-binding sites on the plasma membrane. Binding specificity of the human LH receptor was studied using recombinant human CG, LH, and FSH secreted by CHO cells transfected with the respective genes. Human CG and LH displaced [125I]hCG binding with an ED50 of 4.3 and 4.8 ng/ml, respectively. In contrast, recombinant FSH was not effective. Treatment of transfected cells with recombinant gonadotropins also induced dose-dependent increases in extracellular cAMP production (hCG = LH much greater than FSH; ED50 25, 10, and greater than 3000 ng/ml). Although equine, rat, and ovine LH as well as equine CG competed effectively for rat testicular LH receptor binding, these hormones were unable to displace [125I]hCG binding to the human receptor, suggesting evolutionary changes in receptor binding specificity and the importance of using human receptors for clinical studies. Thus, the cloning and expression of the human LH receptor cDNA allowed analysis of interactions between human LH receptor and gonadotropins from diverse species. The present work should provide the basis for future design of therapeutic agents capable of interacting with the human receptor and for understanding the structural basis for LH receptor binding to different gonadotropins. | 1922095
|