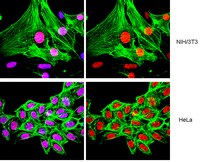
Methylation of histone H3 lysine 9 occurs during translation.
Rivera, C; Saavedra, F; Alvarez, F; Díaz-Celis, C; Ugalde, V; Li, J; Forné, I; Gurard-Levin, ZA; Almouzni, G; Imhof, A; Loyola, A
Nucleic acids research
43
9097-106
2015
Abstract anzeigen
Histone post-translational modifications are key contributors to chromatin structure and function, and participate in the maintenance of genome stability. Understanding the establishment and maintenance of these marks, along with their misregulation in pathologies is thus a major focus in the field. While we have learned a great deal about the enzymes regulating histone modifications on nucleosomal histones, much less is known about the mechanisms establishing modifications on soluble newly synthesized histones. This includes methylation of lysine 9 on histone H3 (H3K9), a mark that primes the formation of heterochromatin, a critical chromatin landmark for genome stability. Here, we report that H3K9 mono- and dimethylation is imposed during translation by the methyltransferase SetDB1. We discuss the importance of these results in the context of heterochromatin establishment and maintenance and new therapeutic opportunities in pathologies where heterochromatin is perturbed. | | | 26405197
 |
The complex pattern of epigenomic variation between natural yeast strains at single-nucleosome resolution.
Filleton, F; Chuffart, F; Nagarajan, M; Bottin-Duplus, H; Yvert, G
Epigenetics & chromatin
8
26
2015
Abstract anzeigen
Epigenomic studies on humans and model species have revealed substantial inter-individual variation in histone modification profiles. However, the pattern of this variation has not been precisely characterized, particularly regarding which genomic features are enriched for variability and whether distinct histone marks co-vary synergistically. Yeast allows us to investigate intra-species variation at high resolution while avoiding other sources of variation, such as cell type or subtype.We profiled histone marks H3K4me3, H3K9ac, H3K14ac, H4K12ac and H3K4me1 in three unrelated wild strains of Saccharomyces cerevisiae at single-nucleosome resolution and analyzed inter-strain differences statistically. All five marks varied significantly at specific loci, but to different extents. The number of nucleosomes varying for a given mark between two strains ranged from 20 to several thousands; +1 nucleosomes were significantly less subject to variation. Genes with highly evolvable or responsive expression showed higher variability; however, the variation pattern could not be explained by known transcriptional differences between the strains. Synergistic variation of distinct marks was not systematic, with surprising differences between functionally related H3K9ac and H3K14ac. Interestingly, H3K14ac differences that persisted through transient hyperacetylation were supported by H3K4me3 differences, suggesting stabilization via cross talk.Quantitative variation of histone marks among S. cerevisiae strains is abundant and complex. Its relation to functional characteristics is modular and seems modest, with partial association with gene expression divergences, differences between functionally related marks and partial co-variation between marks that may confer stability. Thus, the specific context of studies, such as which precise marks, individuals and genomic loci are investigated, is primordial in population epigenomics studies. The complexity found in this pilot survey in yeast suggests that high complexity can be anticipated among higher eukaryotes, including humans. | | | 26229551
|
TELP, a sensitive and versatile library construction method for next-generation sequencing.
Peng, X; Wu, J; Brunmeir, R; Kim, SY; Zhang, Q; Ding, C; Han, W; Xie, W; Xu, F
Nucleic acids research
43
e35
2015
Abstract anzeigen
Next-generation sequencing has been widely used for the genome-wide profiling of histone modifications, transcription factor binding and gene expression through chromatin immunoprecipitated DNA sequencing (ChIP-seq) and cDNA sequencing (RNA-seq). Here, we describe a versatile library construction method that can be applied to both ChIP-seq and RNA-seq on the widely used Illumina platforms. Standard methods for ChIP-seq library construction require nanograms of starting DNA, substantially limiting its application to rare cell types or limited clinical samples. By minimizing the DNA purification steps that cause major sample loss, our method achieved a high sensitivity in ChIP-seq library preparation. Using this method, we achieved the following: (i) generated high-quality epigenomic and transcription factor-binding maps using ChIP-seq for murine adipocytes; (ii) successfully prepared a ChIP-seq library from as little as 25 pg of starting DNA; (iii) achieved paired-end sequencing of the ChIP-seq libraries; (iv) systematically profiled gene expression dynamics during murine adipogenesis using RNA-seq and (v) preserved the strand specificity of the transcripts in RNA-seq. Given its sensitivity and versatility in both double-stranded and single-stranded DNA library construction, this method has wide applications in genomic, epigenomic, transcriptomic and interactomic studies. | | Mouse | 25223787
|
Characterization of BRD4 during mammalian postmeiotic sperm development.
Bryant, JM; Donahue, G; Wang, X; Meyer-Ficca, M; Luense, LJ; Weller, AH; Bartolomei, MS; Blobel, GA; Meyer, RG; Garcia, BA; Berger, SL
Molecular and cellular biology
35
1433-48
2015
Abstract anzeigen
During spermiogenesis, the postmeiotic phase of mammalian spermatogenesis, transcription is progressively repressed as nuclei of haploid spermatids are compacted through a dramatic chromatin reorganization involving hyperacetylation and replacement of most histones with protamines. Although BRDT functions in transcription and histone removal in spermatids, it is unknown whether other BET family proteins play a role. Immunofluorescence of spermatogenic cells revealed BRD4 in a ring around the nuclei of spermatids containing hyperacetylated histones. The ring lies directly adjacent to the acroplaxome, the cytoskeletal base of the acrosome, previously linked to chromatin reorganization. The BRD4 ring does not form in acrosomal mutant mice. Chromatin immunoprecipitation followed by sequencing in spermatids revealed enrichment of BRD4 and acetylated histones at the promoters of active genes. BRD4 and BRDT show distinct and synergistic binding patterns, with a pronounced enrichment of BRD4 at spermatogenesis-specific genes. Direct association of BRD4 with acetylated H4 decreases in late spermatids as acetylated histones are removed from the condensing nucleus in a wave following the progressing acrosome. These data provide evidence of a prominent transcriptional role for BRD4 and suggest a possible removal mechanism for chromatin components from the genome via the progressing acrosome as transcription is repressed and chromatin is compacted during spermiogenesis. | Immunofluorescence | | 25691659
|
H4K12ac is regulated by estrogen receptor-alpha and is associated with BRD4 function and inducible transcription.
Nagarajan, S; Benito, E; Fischer, A; Johnsen, SA
Oncotarget
6
7305-17
2015
Abstract anzeigen
Hormone-dependent gene expression requires dynamic and coordinated epigenetic changes. Estrogen receptor-positive (ER+) breast cancer is particularly dependent upon extensive chromatin remodeling and changes in histone modifications for the induction of hormone-responsive gene expression. Our previous studies established an important role of bromodomain-containing protein-4 (BRD4) in promoting estrogen-regulated transcription and proliferation of ER+ breast cancer cells. Here, we investigated the association between genome-wide occupancy of histone H4 acetylation at lysine 12 (H4K12ac) and BRD4 in the context of estrogen-induced transcription. Similar to BRD4, we observed that H4K12ac occupancy increases near the transcription start sites (TSS) of estrogen-induced genes as well as at distal ERα binding sites in an estrogen-dependent manner. Interestingly, H4K12ac occupancy highly correlates with BRD4 binding and enhancer RNA production on ERα-positive enhancers. Consistent with an importance in estrogen-induced gene transcription, H4K12ac occupancy globally increased in ER-positive cells relative to ER-negative cells and these levels were further increased by estrogen treatment in an ERα-dependent manner. Together, these findings reveal a strong correlation between H4K12ac and BRD4 occupancy with estrogen-dependent gene transcription and further suggest that modulators of H4K12ac and BRD4 may serve as new therapeutic targets for hormone-dependent cancers. | | | 25788266
|
Uncoupling transcription from covalent histone modification.
Zhang, H; Gao, L; Anandhakumar, J; Gross, DS
PLoS genetics
10
e1004202
2014
Abstract anzeigen
It is widely accepted that transcriptional regulation of eukaryotic genes is intimately coupled to covalent modifications of the underlying chromatin template, and in certain cases the functional consequences of these modifications have been characterized. Here we present evidence that gene activation in the silent heterochromatin of the yeast Saccharomyces cerevisiae can occur in the context of little, if any, covalent histone modification. Using a SIR-regulated heat shock-inducible transgene, hsp82-2001, and a natural drug-inducible subtelomeric gene, YFR057w, as models we demonstrate that substantial transcriptional induction (greater than 200-fold) can occur in the context of restricted histone loss and negligible levels of H3K4 trimethylation, H3K36 trimethylation and H3K79 dimethylation, modifications commonly linked to transcription initiation and elongation. Heterochromatic gene activation can also occur with minimal H3 and H4 lysine acetylation and without replacement of H2A with the transcription-linked variant H2A.Z. Importantly, absence of histone modification does not stem from reduced transcriptional output, since hsp82-ΔTATA, a euchromatic promoter mutant lacking a TATA box and with threefold lower induced transcription than heterochromatic hsp82-2001, is strongly hyperacetylated in response to heat shock. Consistent with negligible H3K79 dimethylation, dot1Δ cells lacking H3K79 methylase activity show unimpeded occupancy of RNA polymerase II within activated heterochromatic promoter and coding regions. Our results indicate that large increases in transcription can be observed in the virtual absence of histone modifications often thought necessary for gene activation. | | | 24722509
|
Deletion of a conserved cis-element in the Ifng locus highlights the role of acute histone acetylation in modulating inducible gene transcription.
Balasubramani, A; Winstead, CJ; Turner, H; Janowski, KM; Harbour, SN; Shibata, Y; Crawford, GE; Hatton, RD; Weaver, CT
PLoS genetics
10
e1003969
2014
Abstract anzeigen
Differentiation-dependent regulation of the Ifng cytokine gene locus in T helper (Th) cells has emerged as an excellent model for functional study of distal elements that control lineage-specific gene expression. We previously identified a cis-regulatory element located 22 kb upstream of the Ifng gene (Conserved Non-coding Sequence -22, or CNS-22) that is a site for recruitment of the transcription factors T-bet, Runx3, NF-κB and STAT4, which act to regulate transcription of the Ifng gene in Th1 cells. Here, we report the generation of mice with a conditional deletion of CNS-22 that has enabled us to define the epigenetic and functional consequences of its absence. Deletion of CNS-22 led to a defect in induction of Ifng by the cytokines IL-12 and IL-18, with a more modest effect on induction via T-cell receptor activation. To better understand how CNS-22 and other Ifng CNSs regulated Ifng transcription in response to these distinct stimuli, we examined activation-dependent changes in epigenetic modifications across the extended Ifng locus in CNS-22-deficient T cells. We demonstrate that in response to both cytokine and TCR driven activation signals, CNS-22 and other Ifng CNSs recruit increased activity of histone acetyl transferases (HATs) that transiently enhance levels of histones H3 and H4 acetylation across the extended Ifng locus. We also demonstrate that activation-responsive increases in histone acetylation levels are directly linked to the ability of Ifng CNSs to acutely enhance Pol II recruitment to the Ifng promoter. Finally, we show that impairment in IL-12+IL-18 dependent induction of Ifng stems from the importance of CNS-22 in coordinating locus-wide levels of histone acetylation in response to these cytokines. These findings identify a role for acute histone acetylation in the enhancer function of distal conserved cis-elements that regulate of Ifng gene expression. | | | 24415943
|
The molecular topography of silenced chromatin in Saccharomyces cerevisiae.
Thurtle, DM; Rine, J
Genes & development
28
245-58
2014
Abstract anzeigen
Heterochromatin imparts regional, promoter-independent repression of genes and is epigenetically heritable. Understanding how silencing achieves this regional repression is a fundamental problem in genetics and development. Current models of yeast silencing posit that Sir proteins, recruited by transcription factors bound to the silencers, spread throughout the silenced region. To test this model directly at high resolution, we probed the silenced chromatin architecture by chromatin immunoprecipitation (ChIP) followed by next-generation sequencing (ChIP-seq) of Sir proteins, histones, and a key histone modification, H4K16-acetyl. These analyses revealed that Sir proteins are strikingly concentrated at and immediately adjacent to the silencers, with lower levels of enrichment over the promoters at HML and HMR, the critical targets for transcriptional repression. The telomeres also showed discrete peaks of Sir enrichment yet a continuous domain of hypoacetylated histone H4K16. Surprisingly, ChIP-seq of cross-linked chromatin revealed a distribution of nucleosomes at silenced loci that was similar to Sir proteins, whereas native nucleosome maps showed a regular distribution throughout silenced loci, indicating that cross-linking captured a specialized chromatin organization imposed by Sir proteins. This specialized chromatin architecture observed in yeast informs the importance of a steric contribution to regional repression in other organisms. | | | 24493645
|
Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components.
Alabert, C; Bukowski-Wills, JC; Lee, SB; Kustatscher, G; Nakamura, K; de Lima Alves, F; Menard, P; Mejlvang, J; Rappsilber, J; Groth, A
Nature cell biology
16
281-93
2014
Abstract anzeigen
To maintain genome function and stability, DNA sequence and its organization into chromatin must be duplicated during cell division. Understanding how entire chromosomes are copied remains a major challenge. Here, we use nascent chromatin capture (NCC) to profile chromatin proteome dynamics during replication in human cells. NCC relies on biotin-dUTP labelling of replicating DNA, affinity purification and quantitative proteomics. Comparing nascent chromatin with mature post-replicative chromatin, we provide association dynamics for 3,995 proteins. The replication machinery and 485 chromatin factors such as CAF-1, DNMT1 and SUV39h1 are enriched in nascent chromatin, whereas 170 factors including histone H1, DNMT3, MBD1-3 and PRC1 show delayed association. This correlates with H4K5K12diAc removal and H3K9me1 accumulation, whereas H3K27me3 and H3K9me3 remain unchanged. Finally, we combine NCC enrichment with experimentally derived chromatin probabilities to predict a function in nascent chromatin for 93 uncharacterized proteins, and identify FAM111A as a replication factor required for PCNA loading. Together, this provides an extensive resource to understand genome and epigenome maintenance. | | | 24561620
|
Lunasin sensitivity in non-small cell lung cancer cells is linked to suppression of integrin signaling and changes in histone acetylation.
Inaba, J; McConnell, EJ; Davis, KR
International journal of molecular sciences
15
23705-24
2014
Abstract anzeigen
Lunasin is a plant derived bioactive peptide with both cancer chemopreventive and therapeutic activity. We recently showed lunasin inhibits non-small cell lung cancer (NSCLC) cell proliferation in a cell-line-specific manner. We now compared the effects of lunasin treatment of lunasin-sensitive (H661) and lunasin-insensitive (H1299) NSCLC cells with respect to lunasin uptake, histone acetylation and integrin signaling. Both cell lines exhibited changes in histone acetylation, with H661 cells showing a unique increase in H4K16 acetylation. Proximity ligation assays demonstrated lunasin interacted with integrins containing αv, α5, β1 and β3 subunits to a larger extent in the H661 compared to H1299 cells. Moreover, lunasin specifically disrupted the interaction of β1 and β3 subunits with the downstream signaling components phosphorylated Focal Adhesion Kinase (pFAK), Kindlin and Intergrin Linked Kinase in H661 cells. Immunoblot analyses demonstrated lunasin treatment of H661 resulted in reduced levels of pFAK, phosphorylated Akt and phosphorylated ERK1/2 whereas no changes were observed in H1299 cells. Silencing of αv expression in H661 cells confirmed signaling through integrins containing αv is essential for proliferation. Moreover, lunasin was unable to further inhibit proliferation in αv-silenced H661 cells. This indicates antagonism of integrin signaling via αv-containing integrins is an important component of lunasin's mechanism of action. | Western Blotting | | 25530619
|