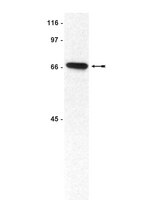
EXO1 is critical for embryogenesis and the DNA damage response in mice with a hypomorphic Nbs1 allele.
Rein, K; Yanez, DA; Terré, B; Palenzuela, L; Aivio, S; Wei, K; Edelmann, W; Stark, JM; Stracker, TH
Nucleic acids research
43
7371-87
2015
Abstract anzeigen
The maintenance of genome stability is critical for the suppression of diverse human pathologies that include developmental disorders, premature aging, infertility and predisposition to cancer. The DNA damage response (DDR) orchestrates the appropriate cellular responses following the detection of lesions to prevent genomic instability. The MRE11 complex is a sensor of DNA double strand breaks (DSBs) and plays key roles in multiple aspects of the DDR, including DNA end resection that is critical for signaling and DNA repair. The MRE11 complex has been shown to function both upstream and in concert with the 5'-3' exonuclease EXO1 in DNA resection, but it remains unclear to what extent EXO1 influences DSB responses independently of the MRE11 complex. Here we examine the genetic relationship of the MRE11 complex and EXO1 during mammalian development and in response to DNA damage. Deletion of Exo1 in mice expressing a hypomorphic allele of Nbs1 leads to severe developmental impairment, embryonic death and chromosomal instability. While EXO1 plays a minimal role in normal cells, its loss strongly influences DNA replication, DNA repair, checkpoint signaling and damage sensitivity in NBS1 hypomorphic cells. Collectively, our results establish a key role for EXO1 in modulating the severity of hypomorphic MRE11 complex mutations. | | 26160886
 |
Evaluation of novel imidazotetrazine analogues designed to overcome temozolomide resistance and glioblastoma regrowth.
Ramirez, YP; Mladek, AC; Phillips, RM; Gynther, M; Rautio, J; Ross, AH; Wheelhouse, RT; Sakaria, JN
Molecular cancer therapeutics
14
111-9
2015
Abstract anzeigen
The cellular responses to two new temozolomide (TMZ) analogues, DP68 and DP86, acting against glioblastoma multiforme (GBM) cell lines and primary culture models are reported. Dose-response analysis of cultured GBM cells revealed that DP68 is more potent than DP86 and TMZ and that DP68 was effective even in cell lines resistant to TMZ. On the basis of a serial neurosphere assay, DP68 inhibits repopulation of these cultures at low concentrations. The efficacy of these compounds was independent of MGMT and MMR functions. DP68-induced interstrand DNA cross-links were demonstrated with H2O2-treated cells. Furthermore, DP68 induced a distinct cell-cycle arrest with accumulation of cells in S phase that is not observed for TMZ. Consistent with this biologic response, DP68 induces a strong DNA damage response, including phosphorylation of ATM, Chk1 and Chk2 kinases, KAP1, and histone variant H2AX. Suppression of FANCD2 expression or ATR expression/kinase activity enhanced antiglioblastoma effects of DP68. Initial pharmacokinetic analysis revealed rapid elimination of these drugs from serum. Collectively, these data demonstrate that DP68 is a novel and potent antiglioblastoma compound that circumvents TMZ resistance, likely as a result of its independence from MGMT and mismatch repair and its capacity to cross-link strands of DNA. | | 25351918
|
CHK1-driven histone H3.3 serine 31 phosphorylation is important for chromatin maintenance and cell survival in human ALT cancer cells.
Chang, FT; Chan, FL; R McGhie, JD; Udugama, M; Mayne, L; Collas, P; Mann, JR; Wong, LH
Nucleic acids research
43
2603-14
2015
Abstract anzeigen
Human ALT cancers show high mutation rates in ATRX and DAXX. Although it is well known that the absence of ATRX/DAXX disrupts H3.3 deposition at heterochromatin, its impact on H3.3 deposition and post-translational modification in the global genome remains unclear. Here, we explore the dynamics of phosphorylated H3.3 serine 31 (H3.3S31ph) in human ALT cancer cells. While H3.3S31ph is found only at pericentric satellite DNA repeats during mitosis in most somatic human cells, a high level of H3.3S31ph is detected on the entire chromosome in ALT cells, attributable to an elevated CHK1 activity in these cells. Drug inhibition of CHK1 activity during mitosis and expression of mutant H3.3S31A in these ALT cells result in a decrease in H3.3S31ph levels accompanied with increased levels of phosphorylated H2AX serine 139 on chromosome arms and at the telomeres. Furthermore, the inhibition of CHK1 activity in these cells also reduces cell viability. Our findings suggest a novel role of CHK1 as an H3.3S31 kinase, and that CHK1-mediated H3.3S31ph plays an important role in the maintenance of chromatin integrity and cell survival in ALT cancer cells. | | 25690891
|
Mir-23a induces telomere dysfunction and cellular senescence by inhibiting TRF2 expression.
Luo, Z; Feng, X; Wang, H; Xu, W; Zhao, Y; Ma, W; Jiang, S; Liu, D; Huang, J; Songyang, Z
Aging cell
14
391-9
2015
Abstract anzeigen
Telomeric repeat binding factor 2 (TRF2) is essential for telomere maintenance and has been implicated in DNA damage response and aging. Telomere dysfunction induced by TRF2 inhibition can accelerate cellular senescence in human fibroblasts. While previous work has demonstrated that a variety of factors can regulate TRF2 expression transcriptionally and post-translationally, whether microRNAs (miRNAs) also participate in post-transcriptionally modulating TRF2 levels remains largely unknown. To better understand the regulatory pathways that control TRF2, we carried out a large-scale luciferase reporter screen using a miRNA expression library and identified four miRNAs that could target human TRF2 and significantly reduce the level of endogenous TRF2 proteins. In particular, our data revealed that miR-23a could directly target the 3' untranslated region (3'UTR) of TRF2. Overexpression of miR-23a not only reduced telomere-bound TRF2 and increased telomere dysfunction-induced foci (TIFs), but also accelerated senescence of human fibroblast cells, which could be rescued by ectopically expressed TRF2. Our findings demonstrate that TRF2 is a specific target of miR-23a, and uncover a previously unknown role for miR-23a in telomere regulation and cellular senescence. | | 25753893
|
Aberrant topoisomerase-1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes.
Katyal, S; Lee, Y; Nitiss, KC; Downing, SM; Li, Y; Shimada, M; Zhao, J; Russell, HR; Petrini, JH; Nitiss, JL; McKinnon, PJ
Nature neuroscience
17
813-21
2014
Abstract anzeigen
DNA damage is considered to be a prime factor in several spinocerebellar neurodegenerative diseases; however, the DNA lesions underpinning disease etiology are unknown. We observed the endogenous accumulation of pathogenic topoisomerase-1 (Top1)-DNA cleavage complexes (Top1ccs) in murine models of ataxia telangiectasia and spinocerebellar ataxia with axonal neuropathy 1. We found that the defective DNA damage response factors in these two diseases cooperatively modulated Top1cc turnover in a non-epistatic and ATM kinase-independent manner. Furthermore, coincident neural inactivation of ATM and DNA single-strand break repair factors, including tyrosyl-DNA phosphodiesterase-1 or XRCC1, resulted in increased Top1cc formation and excessive DNA damage and neurodevelopmental defects. Notably, direct Top1 poisoning to elevate Top1cc levels phenocopied the neuropathology of the mouse models described above. Our results identify a critical endogenous pathogenic lesion associated with neurodegenerative syndromes arising from DNA repair deficiency, indicating that genome integrity is important for preventing disease in the nervous system. | Western Blotting | 24793032
|
Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe.
Sarbajna, S; Davies, D; West, SC
Genes & development
28
1124-36
2014
Abstract anzeigen
The resolution of recombination intermediates containing Holliday junctions (HJs) is critical for genome maintenance and proper chromosome segregation. Three pathways for HJ processing exist in human cells and involve the following enzymes/complexes: BLM-TopoIIIα-RMI1-RMI2 (BTR complex), SLX1-SLX4-MUS81-EME1 (SLX-MUS complex), and GEN1. Cycling cells preferentially use the BTR complex for the removal of double HJs in S phase, with SLX-MUS and GEN1 acting at temporally distinct phases of the cell cycle. Cells lacking SLX-MUS and GEN1 exhibit chromosome missegregation, micronucleus formation, and elevated levels of 53BP1-positive G1 nuclear bodies, suggesting that defects in chromosome segregation lead to the transmission of extensive DNA damage to daughter cells. In addition, however, we found that the effects of SLX4, MUS81, and GEN1 depletion extend beyond mitosis, since genome instability is observed throughout all phases of the cell cycle. This is exemplified in the form of impaired replication fork movement and S-phase progression, endogenous checkpoint activation, chromosome segmentation, and multinucleation. In contrast to SLX4, SLX1, the nuclease subunit of the SLX1-SLX4 structure-selective nuclease, plays no role in the replication-related phenotypes associated with SLX4/MUS81 and GEN1 depletion. These observations demonstrate that the SLX1-SLX4 nuclease and the SLX4 scaffold play divergent roles in the maintenance of genome integrity in human cells. | Western Blotting | 24831703
|
Parvovirus-induced depletion of cyclin B1 prevents mitotic entry of infected cells.
Adeyemi, RO; Pintel, DJ
PLoS pathogens
10
e1003891
2014
Abstract anzeigen
Parvoviruses halt cell cycle progression following initiation of their replication during S-phase and continue to replicate their genomes for extended periods of time in arrested cells. The parvovirus minute virus of mice (MVM) induces a DNA damage response that is required for viral replication and induction of the S/G2 cell cycle block. However, p21 and Chk1, major effectors typically associated with S-phase and G2-phase cell cycle arrest in response to diverse DNA damage stimuli, are either down-regulated, or inactivated, respectively, during MVM infection. This suggested that parvoviruses can modulate cell cycle progression by another mechanism. In this work we show that the MVM-induced, p21- and Chk1-independent, cell cycle block proceeds via a two-step process unlike that seen in response to other DNA-damaging agents or virus infections. MVM infection induced Chk2 activation early in infection which led to a transient S-phase block associated with proteasome-mediated CDC25A degradation. This step was necessary for efficient viral replication; however, Chk2 activation and CDC25A loss were not sufficient to keep infected cells in the sustained G2-arrested state which characterizes this infection. Rather, although the phosphorylation of CDK1 that normally inhibits entry into mitosis was lost, the MVM induced DDR resulted first in a targeted mis-localization and then significant depletion of cyclin B1, thus directly inhibiting cyclin B1-CDK1 complex function and preventing mitotic entry. MVM infection thus uses a novel strategy to ensure a pseudo S-phase, pre-mitotic, nuclear environment for sustained viral replication. | | 24415942
|
Conditional inactivation of the DNA damage response gene Hus1 in mouse testis reveals separable roles for components of the RAD9-RAD1-HUS1 complex in meiotic chromosome maintenance.
Lyndaker, AM; Lim, PX; Mleczko, JM; Diggins, CE; Holloway, JK; Holmes, RJ; Kan, R; Schlafer, DH; Freire, R; Cohen, PE; Weiss, RS
PLoS genetics
9
e1003320
2013
Abstract anzeigen
The RAD9-RAD1-HUS1 (9-1-1) complex is a heterotrimeric PCNA-like clamp that responds to DNA damage in somatic cells by promoting DNA repair as well as ATR-dependent DNA damage checkpoint signaling. In yeast, worms, and flies, the 9-1-1 complex is also required for meiotic checkpoint function and efficient completion of meiotic recombination; however, since Rad9, Rad1, and Hus1 are essential genes in mammals, little is known about their functions in mammalian germ cells. In this study, we assessed the meiotic functions of 9-1-1 by analyzing mice with germ cell-specific deletion of Hus1 as well as by examining the localization of RAD9 and RAD1 on meiotic chromosomes during prophase I. Hus1 loss in testicular germ cells resulted in meiotic defects, germ cell depletion, and severely compromised fertility. Hus1-deficient primary spermatocytes exhibited persistent autosomal γH2AX and RAD51 staining indicative of unrepaired meiotic DSBs, synapsis defects, an extended XY body domain often encompassing partial or whole autosomes, and an increase in structural chromosome abnormalities such as end-to-end X chromosome-autosome fusions and ruptures in the synaptonemal complex. Most of these aberrations persisted in diplotene-stage spermatocytes. Consistent with a role for the 9-1-1 complex in meiotic DSB repair, RAD9 localized to punctate, RAD51-containing foci on meiotic chromosomes in a Hus1-dependent manner. Interestingly, RAD1 had a broader distribution that only partially overlapped with RAD9, and localization of both RAD1 and the ATR activator TOPBP1 to the XY body and to unsynapsed autosomes was intact in Hus1 conditional knockouts. We conclude that mammalian HUS1 acts as a component of the canonical 9-1-1 complex during meiotic prophase I to promote DSB repair and further propose that RAD1 and TOPBP1 respond to unsynapsed chromatin through an alternative mechanism that does not require RAD9 or HUS1. | | 23468651
|
The telomere deprotection response is functionally distinct from the genomic DNA damage response.
Cesare, AJ; Hayashi, MT; Crabbe, L; Karlseder, J
Molecular cell
51
141-55
2013
Abstract anzeigen
Loss of chromosome end protection through telomere erosion is a hallmark of aging and senescence. Here we developed an experimental system that mimics physiological telomere deprotection in human cells and discovered that the telomere deprotection response is functionally distinct from the genomic DNA damage response. We found that, unlike genomic breaks, deprotected telomeres that are recognized as DNA damage but remain in the fusion-resistant intermediate state activate differential ataxia telangiectasia mutated (ATM) signaling where CHK2 is not phosphorylated. Also unlike genomic breaks, we found that deprotected telomeres do not contribute to the G2/M checkpoint and are instead passed through cell division to induce p53-dependent G1 arrest in the daughter cells. Telomere deprotection is therefore an epigenetic signal passed between cell generations to ensure that replication-associated telomere-dependent growth arrest occurs in stable diploid G1 phase cells before genome instability can occur. | | 23850488
|
HELQ promotes RAD51 paralogue-dependent repair to avert germ cell loss and tumorigenesis.
Adelman, CA; Lolo, RL; Birkbak, NJ; Murina, O; Matsuzaki, K; Horejsi, Z; Parmar, K; Borel, V; Skehel, JM; Stamp, G; D'Andrea, A; Sartori, AA; Swanton, C; Boulton, SJ
Nature
502
381-4
2013
Abstract anzeigen
Repair of interstrand crosslinks (ICLs) requires the coordinated action of the intra-S-phase checkpoint and the Fanconi anaemia pathway, which promote ICL incision, translesion synthesis and homologous recombination (reviewed in refs 1, 2). Previous studies have implicated the 3'-5' superfamily 2 helicase HELQ in ICL repair in Drosophila melanogaster (MUS301 (ref. 3)) and Caenorhabditis elegans (HELQ-1 (ref. 4)). Although in vitro analysis suggests that HELQ preferentially unwinds synthetic replication fork substrates with 3' single-stranded DNA overhangs and also disrupts protein-DNA interactions while translocating along DNA, little is known regarding its functions in mammalian organisms. Here we report that HELQ helicase-deficient mice exhibit subfertility, germ cell attrition, ICL sensitivity and tumour predisposition, with Helq heterozygous mice exhibiting a similar, albeit less severe, phenotype than the null, indicative of haploinsufficiency. We establish that HELQ interacts directly with the RAD51 paralogue complex BCDX2 and functions in parallel to the Fanconi anaemia pathway to promote efficient homologous recombination at damaged replication forks. Thus, our results reveal a critical role for HELQ in replication-coupled DNA repair, germ cell maintenance and tumour suppression in mammals. | Western Blotting | 24005329
|