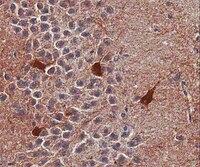
The effect of STAT3 inhibition on status epilepticus and subsequent spontaneous seizures in the pilocarpine model of acquired epilepsy.
Grabenstatter, HL; Del Angel, YC; Carlsen, J; Wempe, MF; White, AM; Cogswell, M; Russek, SJ; Brooks-Kayal, AR
Neurobiology of disease
62
73-85
2014
Show Abstract
Pilocarpine-induced status epilepticus (SE), which results in temporal lobe epilepsy (TLE) in rodents, activates the JAK/STAT pathway. In the current study, we evaluate whether brief exposure to a selective inhibitor of the JAK/STAT pathway (WP1066) early after the onset of SE affects the severity of SE or reduces later spontaneous seizure frequency via inhibition of STAT3-regulated gene transcription. Rats that received systemic WP1066 or vehicle at the onset of SE were continuously video-EEG monitored during SE and for one month to assess seizure frequency over time. Protein and/or mRNA levels for pSTAT3, and STAT3-regulated genes including: ICER, Gabra1, c-myc, mcl-1, cyclin D1, and bcl-xl were evaluated in WP1066 and vehicle-treated rats during stages of epileptogenesis to determine the acute effects of WP1066 administration on SE and chronic epilepsy. WP1066 (two 50mg/kg doses) administered within the first hour after onset of SE results in transient inhibition of pSTAT3 and long-term reduction in spontaneous seizure frequency. WP1066 alters the severity of chronic epilepsy without affecting SE or cell death. Early WP1066 administration reduces known downstream targets of STAT3 transcription 24h after SE including cyclin D1 and mcl-1 levels, known for their roles in cell-cycle progression and cell survival, respectively. These findings uncover a potential effect of the JAK/STAT pathway after brain injury that is physiologically important and may provide a new therapeutic target that can be harnessed for the prevention of epilepsy development and/or progression. | Western Blotting | | 24051278
 |
Synaptic function of nicastrin in hippocampal neurons.
Lee, SH; Sharma, M; Südhof, TC; Shen, J
Proceedings of the National Academy of Sciences of the United States of America
111
8973-8
2014
Show Abstract
Synaptic dysfunction is widely thought to play a key role in the pathogenesis of Alzheimer's disease (AD). Presenilins, the major gene products involved in familial AD, are essential for short- and long-term synaptic plasticity in mature neurons as well as for the survival of cortical neurons during aging. Presenilin and nicastrin are both indispensable components of the γ-secretase complex, but it remains unknown whether presenilin regulates synaptic function in a γ-secretase-dependent or γ-secretase-independent manner and whether nicastrin plays similar roles in central synapses. In the current study, we address these questions using an electrophysiological approach to analyze nicastrin conditional knockout (cKO) mice in the hippocampal Schaffer collateral pathway. In these mice, we found that, even at 2 mo of age, deletion of nicastrin in excitatory neurons of the postnatal forebrain using Cre recombinase expressed under the control of the αCaMKII promoter led to deficits in presynaptic short-term plasticity including paired-pulse facilitation and frequency facilitation. Depletion of Ca(2+) in the endoplasmic reticulum mimics and occludes the presynaptic facilitation deficits in nicastrin cKO mice, suggesting that disrupted intracellular Ca(2+) homeostasis underlies the presynaptic deficits. In addition, NMDA receptor-mediated responses and long-term potentiation induced by theta-burst stimulation were decreased in nicastrin cKO mice at 3 mo but not at 2 mo of age. Together, these findings show that, similar to presenilins, nicastrin plays essential roles in the regulation of short- and long-term synaptic plasticity, highlighting the importance of γ-secretase in the function of mature synapses. | Western Blotting | Mouse | 24889619
|
Cryoloading: introducing large molecules into live synaptosomes.
Nath, AR; Chen, RH; Stanley, EF
Frontiers in cellular neuroscience
8
4
2014
Show Abstract
Neurons communicate with their target cells primarily by the release of chemical transmitters from presynaptic nerve terminals. The study of CNS presynaptic nerve terminals, isolated as synaptosomes (SSMs) has, however, been hampered by the typical small size of these structures that precludes the introduction of non-membrane permeable test substances such as peptides and drugs. We have developed a method to introduce large alien compounds of at least 150 kDa into functional synaptosomes. Purified synaptosomes are frozen in cryo-preserving buffer containing the alien compound. Upon defrosting, many of the SSMs contain the alien compound presumably admitted by bulk buffer-transfer through the surface membranes that crack and reseal during the freeze/thaw cycle. ~80% of the cryoloaded synaptosomes were functional and recycled synaptic vesicles (SVs), as assessed by a standard styryl dye uptake assay. Access of the cryoloaded compound into the cytoplasm and biological activity were confirmed by block of depolarization-induced SV recycling with membrane-impermeant BAPTA (a rapid Ca(2+)-scavenger), or botulinum A light chain (which cleaves the soluble NSF attachment protein receptor (SNARE) protein SNAP25). A major advantage of the method is that loaded frozen synaptosomes can be stored virtually indefinitely for later experimentation. We also demonstrate that individual synaptosome types can be identified by immunostaining of receptors associated with its scab of attached postsynaptic membrane. Thus, cryoloading and scab-staining permits the examination of SV recycling in identified individual CNS presynaptic nerve terminals. | | | 24478628
|
An autocrine γ-aminobutyric acid signaling system exists in pancreatic β-cell progenitors of fetal and postnatal mice.
Feng, MM; Xiang, YY; Wang, S; Lu, WY
International journal of physiology, pathophysiology and pharmacology
5
91-101
2013
Show Abstract
Gamma-aminobutyric acid (GABA) is produced and secreted by adult pancreatic β-cells, which also express GABA receptors mediating autocrine signaling and regulating β-cell proliferation. However, whether the autocrine GABA signaling involves in β-cell progenitor development or maturation remains uncertain. By means of immunohistochemistry we analyzed the expression profiles of the GABA synthesizing enzyme glutamic acid decarboxylase (GAD) and the α1-subunit of type-A GABA receptor (GABAARα1) in the pancreas of mice at embryonic day 15.5 (E15.5), E18.5, postnatal day 1 (P1) and P7. Our data showed that at E15.5 the pancreatic and duodenum homeobox-1 (Pdx1) was expressed in the majority of cells in the developing pancreata. Notably, insulin immunoreactivity was identified in a subpopulation of pancreatic cells with a high level of Pdx1 expression. About 80% of the high-level Pdx-1 expressing cells in the pancreas expressed GAD and GABAARα1 at all pancreatic developmental stages. In contrast, only about 30% of the high-level Pdx-1 expressing cells in the E15.5 pancreas expressed insulin; i.e., a large number of GAD/GABAARα1-expressing cells did not express insulin at this early developmental stage. The expression level of GAD and GABAARα1 increased steadily, and progressively more GAD/GABAARα1-expressing cells expressed insulin in the course of pancreatic development. These results suggest that 1) GABA signaling proteins appear in β-cell progenitors prior to insulin expression; and 2) the increased expression of GABA signaling proteins may be involved in β-cell progenitor maturation. | Immunohistochemistry | | 23750307
|
Altered cortical GABAA receptor composition, physiology, and endocytosis in a mouse model of a human genetic absence epilepsy syndrome.
Zhou, C; Huang, Z; Ding, L; Deel, ME; Arain, FM; Murray, CR; Patel, RS; Flanagan, CD; Gallagher, MJ
The Journal of biological chemistry
288
21458-72
2013
Show Abstract
Patients with generalized epilepsy exhibit cerebral cortical disinhibition. Likewise, mutations in the inhibitory ligand-gated ion channels, GABAA receptors (GABAARs), cause generalized epilepsy syndromes in humans. Recently, we demonstrated that heterozygous knock-out (Hetα1KO) of the human epilepsy gene, the GABAAR α1 subunit, produced absence epilepsy in mice. Here, we determined the effects of Hetα1KO on the expression and physiology of GABAARs in the mouse cortex. We found that Hetα1KO caused modest reductions in the total and surface expression of the β2 subunit but did not alter β1 or β3 subunit expression, results consistent with a small reduction of GABAARs. Cortices partially compensated for Hetα1KO by increasing the fraction of residual α1 subunit on the cell surface and by increasing total and surface expression of α3, but not α2, subunits. Co-immunoprecipitation experiments revealed that Hetα1KO increased the fraction of α1 subunits, and decreased the fraction of α3 subunits, that associated in hybrid α1α3βγ receptors. Patch clamp electrophysiology studies showed that Hetα1KO layer VI cortical neurons exhibited reduced inhibitory postsynaptic current peak amplitudes, prolonged current rise and decay times, and altered responses to benzodiazepine agonists. Finally, application of inhibitors of dynamin-mediated endocytosis revealed that Hetα1KO reduced base-line GABAAR endocytosis, an effect that probably contributes to the observed changes in GABAAR expression. These findings demonstrate that Hetα1KO exerts two principle disinhibitory effects on cortical GABAAR-mediated inhibitory neurotransmission: 1) a modest reduction of GABAAR number and 2) a partial compensation with GABAAR isoforms that possess physiological properties different from those of the otherwise predominant α1βγ GABAARs. | | | 23744069
|
Baicalein reduces β-amyloid and promotes nonamyloidogenic amyloid precursor protein processing in an Alzheimer's disease transgenic mouse model.
Zhang, SQ; Obregon, D; Ehrhart, J; Deng, J; Tian, J; Hou, H; Giunta, B; Sawmiller, D; Tan, J
Journal of neuroscience research
91
1239-46
2013
Show Abstract
Baicalein, a flavonoid isolated from the roots of Scutellaria baicalensis, is known to modulate γ-aminobutyric acid (GABA) type A receptors. Given prior reports demonstrating benefits of GABAA modulation for Alzheimer's disease (AD) treatment, we wished to determine whether this agent might be beneficial for AD. CHO cells engineered to overexpress wild-type amyloid precursor protein (APP), primary culture neuronal cells from AD mice (Tg2576) and AD mice were treated with baicalein. In the cell cultures, baicalein significantly reduced the production of β-amyloid (Aβ) by increasing APP α-processing. These effects were blocked by the GABAA antagonist bicuculline. Likewise, AD mice treated daily with i.p. baicalein for 8 weeks showed enhanced APP α-secretase processing, reduced Aβ production, and reduced AD-like pathology together with improved cognitive performance. Our findings suggest that baicalein promotes nonamyloidogenic processing of APP, thereby reducing Aβ production and improving cognitive performance, by activating GABAA receptors. | Western Blotting | | 23686791
|
Expression of GABAA α2-, β1- and ε-receptors are altered significantly in the lateral cerebellum of subjects with schizophrenia, major depression and bipolar disorder.
Fatemi, SH; Folsom, TD; Rooney, RJ; Thuras, PD
Translational psychiatry
3
e303
2013
Show Abstract
There is abundant evidence that dysfunction of the γ-aminobutyric acid (GABA)ergic signaling system is implicated in the pathology of schizophrenia and mood disorders. Less is known about the alterations in protein expression of GABA receptor subunits in brains of subjects with schizophrenia and mood disorders. We have previously demonstrated reduced expression of GABA(B) receptor subunits 1 and 2 (GABBR1 and GABBR2) in the lateral cerebella of subjects with schizophrenia, bipolar disorder and major depressive disorder. In the current study, we have expanded these studies to examine the mRNA and protein expression of 12 GABA(A) subunit proteins (α1, α2, α3, α5, α6, β1, β2, β3, δ, ε, γ2 and γ3) in the lateral cerebella from the same set of subjects with schizophrenia (N=9-15), bipolar disorder (N=10-15) and major depression (N=12-15) versus healthy controls (N=10-15). We found significant group effects for protein levels of the α2-, β1- and ε-subunits across treatment groups. We also found a significant group effect for mRNA levels of the α1-subunit across treatment groups. New avenues for treatment, such as the use of neurosteroids to promote GABA modulation, could potentially ameliorate GABAergic dysfunction in these disorders. | Western Blotting | Human | 24022508
|
Tracking the expression of excitatory and inhibitory neurotransmission-related proteins and neuroplasticity markers after noise induced hearing loss.
Browne, CJ; Morley, JW; Parsons, CH
PloS one
7
e33272
2012
Show Abstract
Excessive exposure to loud noise can damage the cochlea and create a hearing loss. These pathologies coincide with a range of CNS changes including reorganisation of frequency representation, alterations in the pattern of spontaneous activity and changed expression of excitatory and inhibitory neurotransmitters. Moreover, damage to the cochlea is often accompanied by acoustic disorders such as hyperacusis and tinnitus, suggesting that one or more of these neuronal changes may be involved in these disorders, although the mechanisms remain unknown. We tested the hypothesis that excessive noise exposure increases expression of markers of excitation and plasticity, and decreases expression of inhibitory markers over a 32-day recovery period. Adult rats (n = 25) were monaurally exposed to a loud noise (16 kHz, 1/10(th) octave band pass (115 dB SPL)) for 1-hour, or left as non-exposed controls (n = 5). Animals were euthanased at either 0, 4, 8, 16 or 32 days following acoustic trauma. We used Western Blots to quantify protein levels of GABA(A) receptor subunit α1 (GABA(A)α1), Glutamic-Acid Decarboxylase-67 (GAD-67), N-Methyl-D-Aspartate receptor subunit 2A (NR2A), Calbindin (Calb1) and Growth Associated Protein 43 (GAP-43) in the Auditory Cortex (AC), Inferior Colliculus (IC) and Dorsal Cochlear Nucleus (DCN). Compared to sham-exposed controls, noise-exposed animals had significantly (pless than 0.05): lower levels of GABA(A)α1 in the contralateral AC at day-16 and day-32, lower levels of GAD-67 in the ipsilateral DCN at day-4, lower levels of Calb1 in the ipsilateral DCN at day-0, lower levels of GABA(A)α1 in the ipsilateral AC at day-4 and day-32. GAP-43 was reduced in the ipsilateral AC for the duration of the experiment. These complex fluctuations in protein expression suggests that for at least a month following acoustic trauma the auditory system is adapting to a new pattern of sensory input. | | | 22428005
|
CSPα knockout causes neurodegeneration by impairing SNAP-25 function.
Sharma, M; Burré, J; Bronk, P; Zhang, Y; Xu, W; Südhof, TC
The EMBO journal
31
829-41
2012
Show Abstract
At a synapse, the synaptic vesicle protein cysteine-string protein-α (CSPα) functions as a co-chaperone for the SNARE protein SNAP-25. Knockout (KO) of CSPα causes fulminant neurodegeneration that is rescued by α-synuclein overexpression. The CSPα KO decreases SNAP-25 levels and impairs SNARE-complex assembly; only the latter but not the former is reversed by α-synuclein. Thus, the question arises whether the CSPα KO phenotype is due to decreased SNAP-25 function that then causes neurodegeneration, or due to the dysfunction of multiple as-yet uncharacterized CSPα targets. Here, we demonstrate that decreasing SNAP-25 levels in CSPα KO mice by either KO or knockdown of SNAP-25 aggravated their phenotype. Conversely, increasing SNAP-25 levels by overexpression rescued their phenotype. Inactive SNAP-25 mutants were unable to rescue, showing that the rescue was specific. Under all conditions, the neurodegenerative phenotype precisely correlated with SNARE-complex assembly, indicating that impaired SNARE-complex assembly due to decreased SNAP-25 levels is the ultimate correlate of neurodegeneration. Our findings suggest that the neurodegeneration in CSPα KO mice is primarily produced by defective SNAP-25 function, which causes neurodegeneration by impairing SNARE-complex assembly. | | | 22187053
|
Proteasome Inhibition Alleviates SNARE-Dependent Neurodegeneration.
Sharma, Manu, et al.
Sci Transl Med, 4: 147ra113 (2012)
2012
Show Abstract
Activation of the proteasomal degradation of misfolded proteins has been proposed as a therapeutic strategy for treating neurodegenerative diseases, but it is unclear whether proteasome dysfunction contributes to neurodegeneration. We tested the role of proteasome activity in neurodegeneration developed by mice lacking cysteine string protein-α (CSPα). Unexpectedly, we found that proteasome inhibitors alleviated neurodegeneration in CSPα-deficient mice, reversing impairment of SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor)-complex assembly and extending life span. We tested whether dysfunctional SNARE-complex assembly could contribute to neurodegeneration in Alzheimer's and Parkinson's disease by analyzing postmortem brain tissue from these patients; we found reduced SNARE-complex assembly in the brain tissue samples. Our results suggest that proteasomal activation may not always be beneficial for alleviating neurodegeneration and that blocking the proteasome may represent a potential therapeutic avenue for treating some forms of neurodegenerative disease. | | | 22896677
|